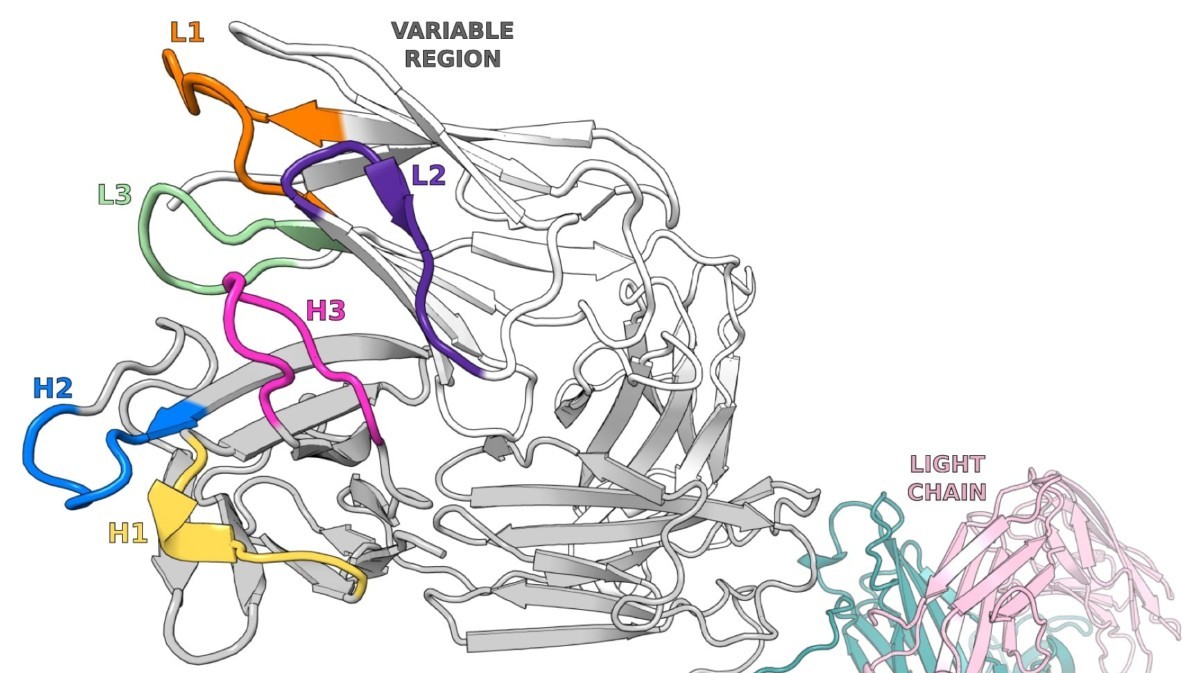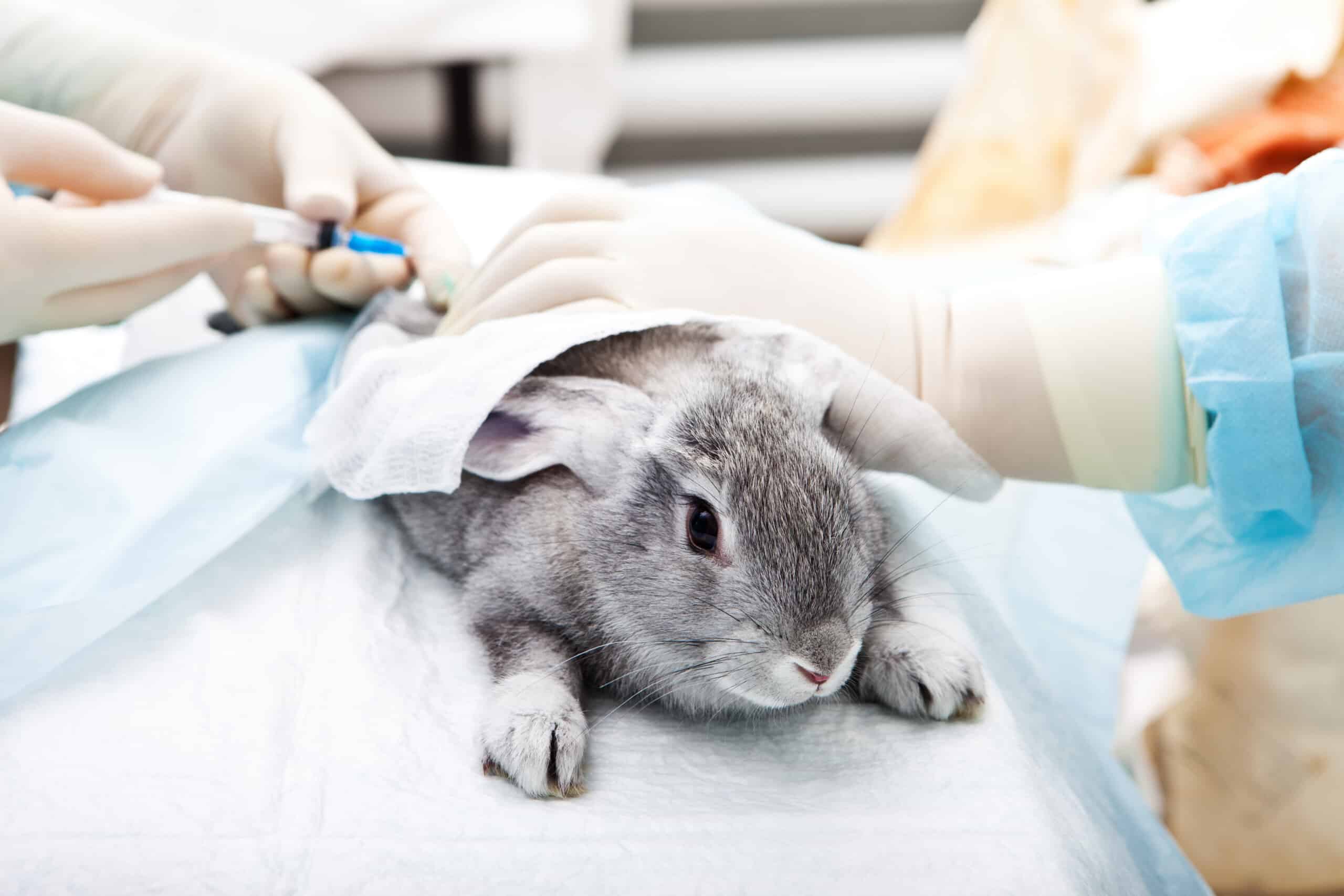
Animal Testing in Biomedical Research and New Approach Methodologies (NAMs)
Written By: Monica Mioduszewski Published: April 16, 2026 Contents Introduction How many animals are used in research? Which animals are used for testing? Why animal models remain in use Limitations of Animal Models New Approach Methodologies (NAMs): potential solutions to animal testing A persistent challenge: antibody generation Polyclonal Antibody Sequencing [...]