 Written by:
Written by:
Vanessa Yoon Calvelo, PhD
Published: July 21, 2022
Mass spectrometry-based approaches for structural analysis of proteins
Characterization of proteins and protein complexes is a major keystone of structural biology. As our understanding of cellular processes continues to evolve from simple pathways to complicated networks, our need for advanced analytical methods is quite apparent. Mass spectrometry (MS)-based structural approaches can be used to study protein conformational changes and dynamics, protein motion/flexibility, ligand-protein binding, and protein-protein interfaces. Though there are other established structural, analytical methods, such as those listed in Table 1, one advantage for MS-based techniques is their capacity to characterize ‘difficult’ proteins that otherwise would be challenging to study. One such technique is hydrogen-deuterium exchange mass spectrometry (HDX-MS), in which protein characterization relies on the process of protein deuteration to distinguish amide hydrogens participating in secondary structures.
The scope of this article will focus on the applications of HDX-MS for the analysis of different ‘difficult’ protein types. For an overview of the HDX-MS method and underlying theory, please refer to our article: What is HDX-MS Epitope Mapping?. For a more in-depth review on HDX-MS and its applications, please refer to the publication by Narang et al. (2020).
Table 1. Analytical methods for studying structures of proteins and protein complexes (adapted from Xiao et al. (2018)).
![]()
HDX-MS Applications for ‘Difficult’ Proteins
As stated above, the capacity to characterize ‘difficult’ proteins offers HDX-MS a preferential edge as a high-resolution, structural, analytical technique. The functionality of HDX-MS to obtain structural information on some of these proteins and protein complexes will be discussed below (Figure 1).
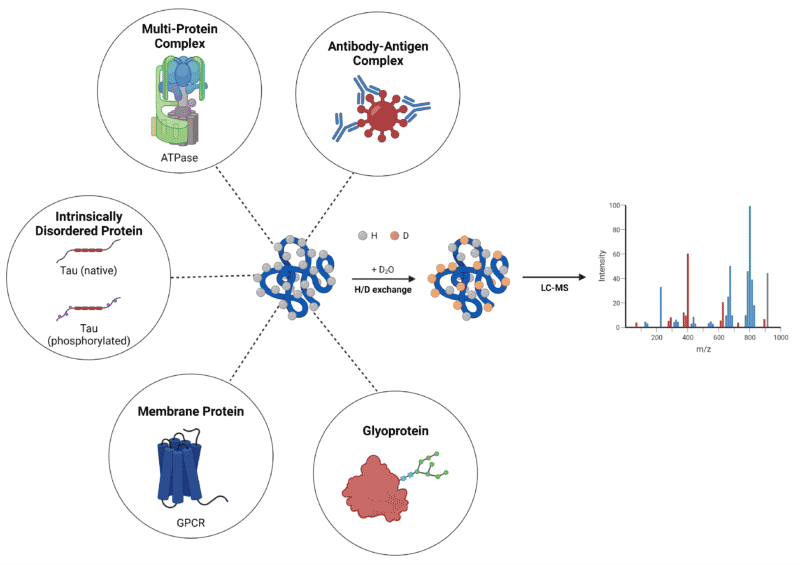
Figure 1. Summary of ‘difficult’ protein types characterized by HDX-MS.
Antibody-Antigen Complexes
A burgeoning industrial application of HDX-MS is the characterization of antibody-antigen complexes to reveal the antibody binding region on the antigen surface. This method, also known as epitope mapping, can provide valuable insights into the dynamic conformation of the antibody, as well as any changes induced by the presence of the antigen binding partner.
HDX-MS has already emerged as a method of choice for mapping epitopes of monoclonal antibodies (mAbs). Thus far, epitope mapping studies almost exclusively focus on mAbs – one example is the mapping of two mAbs against diphtheria toxin, revealing their distinct neutralizing mechanisms.
The mapping of polyclonal antibodies (pAbs), on the other hand, is still emerging due to the heterogeneity of pAb samples. Very few methods for epitope mapping can tolerate this complexity; however, since HDX-MS is typically amenable to difficult samples, it has the potential to be optimized for pAb characterization. Examples of HDX-MS applications for pAb analysis include: 1) the epitope mapping of polyclonal antibodies against the Neisseria meningitidis vaccine antigen, factor H-binding protein (fHbp); and 2) the epitope mapping of polyclonal antibodies against the cashew allergen antigen, Ana o 2.
Multi-Protein Complexes
Many cellular processes require structural assemblies of large complexes that are the result of interactions between two or more associated proteins. The nature of their large size often poses a challenge in studying these protein complexes with standard methods, such as X-ray crystallography (XRC) and nuclear magnetic resonance (NMR) spectroscopy – this is where applications of HDX-MS are particularly strong. With advancements in liquid chromatography (LC) systems and MS resolution, large protein complexes can be conformationally characterized using bottom-up HDX-MS.
One example is the characterization of the 525-kDa, membrane-embedded FOF1 ATP synthase complex that is responsible for ATP synthesis and hydrolysis. FOF1 typically exists in three conformations: 1) an inhibited conformation when bound to ADP; 2) a catalytically active conformation when the inner-membrane proton motive force is high; and 3) a working conformation when carbonyl-cyanide-4-(trifluoromethoxy) phenylhydrazone prevents proton build-up in the membrane. HDX-MS was utilized to compare all three conformations of the FOF1 complex, revealing functional insights into this biological motor and many others like it.
Intrinsically Disordered Proteins
Many cellular processes involve the interactions of intrinsically disordered proteins (IDPs) – proteins that lack a well-defined tertiary structure in its native state – or protein hybrids that contain both ordered and intrinsically disordered regions (IDRs). Unhindered by energetic constraints, IDPs are highly dynamic in solution and their structure-independent function stems from their ability to interconvert between multiple transient conformations or residual structures.
The dynamic nature of IDPs and IDRs complicates their structural characterization. Difficulties in protein crystallization renders them unsuitable for analysis by XRC. Elucidating copopulated IDP states due to fluctuating orientations of the mobile domains remains challenging with cryo-electron microscopy. NMR hurdles include difficulty in distinguishing between multiple coexisting conformations due to rapid interconversions, as well as protein size limitations and the requirement of high sample concentrations. Therefore, HDX-MS can have a leading advantage in IDP characterization where the folded protein form is not the preferred or dominant state.
HDX-MS can accommodate the millisecond-labeling time to detect transiently populated IDP states. One example is the application of HDX-MS for the characterization of the intrinsically disordered, amyloidogenic tau protein. Tau, a microtubule-associated protein that is often a biomarker for neurodegenerative diseases including Alzheimer’s disease, contains a microtubule-binding region, as well as two hexapeptide motifs H1 (275VQIINK280) and H2 (306VQIVYK311). Using HDX-MS, specific structural changes – a release in its global fold, the development of new intramolecular interactions in the microtubule-binding region, as well as increased exposure of H2 – were characterized as tau transitions from its residual structure to its pathogenic state.
Membrane Proteins
Membrane proteins are another protein group required for many cellular processes and are often major targets for drug discovery and design. As a result, understanding their conformational dynamics is crucial; however, membrane proteins are among the most challenging to study in structural biology. Their stability and aggregation in solution makes them particularly difficult to characterize using standard methods, such as XRC and NMR. HDX-MS, on the other hand, has been extensively used to characterize the conformations of membrane proteins, including G-protein coupled receptors in their unbound and ligand-bound states. One pioneering example is the characterization of β2-Adrenergic GPCR (β2AR) by HDX-MS, comparing its unbound state to its bound state with the inverse agonist carazolol.
When analyzing membrane proteins, sample preparation requires a solubilization step typically carried out using micelles, bicelles, nanodiscs, or other types of detergents. Though the applicability of HDX-MS covers membrane proteins, the presence of lipids, nanodiscs, and detergents can affect the performance of LC-MS systems and will require parameter optimization.
Glycosylated Proteins, or Glycoproteins
Protein glycosylation, one of the most prominent types of post-translational modifications, involves the covalent attachment of single sugars or glycans (N-linked or O-linked glycans) to select amino acid residues of target proteins. At least half of the protein population in humans is glycosylated, having implications in many cellular processes. Glycoproteins typically have several glycosylation sites, with each site occupied by different glycans, resulting in a wide range of glycoforms of a single protein. This complexity often poses difficulties in the structural analysis of glycoproteins – flexibility of the glycan chain can prevent crystallization limiting X-ray diffraction methods, and glycan heterogeneity can cause significant signal interference in NMR analysis.
HDX-MS is inherently suitable to study multifaceted protein states. However, several challenges still exist with glycoprotein characterization, including: 1) limited digestion of the glycosylated protein due to steric hindrance from the glycans; 2) glycopeptide signal spread due to the coexistence of several glycoforms; 3) retention of deuterium by acetamido groups in the glycan; and 4) limited binding of peptides to the LC column due to the hydrophilic nature of glycans. However, with adjustments and optimization of procedural workflows, HDX-MS applications are continuing to expand in the structural analysis of glycoproteins. One example is the employment of PNGase A that deglycosylates proteins under acidic pH and low temperature, reducing the complexity of the glycoprotein sample.
HDX-MS – A powerful analytical technique for protein characterization
As highlighted above, the versatility of HDX-MS enables the study of protein folding intermediates, rarely populated protein states, large macromolecular assemblies, and protein-protein interactions. However, this is not to say that this technique is without its challenges. Each unique protein requires optimization of the workflow and the peripheral factors that influence deuterium uptake, including temperature, pH (pD), and ionic strength of the buffer solution. Challenges aside, HDX-MS has provided essential knowledge on some of the most ‘difficult’ biological targets (Figure 1). With further advancements and automation of this MS-based technique, HDX systems will only become more user friendly with broader applications.
Rapid HDX-MS
At Rapid Novor, we provide HDX-MS epitope mapping services as an efficient and effective solution to characterizing antibody-antigen interactions. We provide a bottom-up MS-based workflow to rapidly and accurately determine linear, conformational, and structural epitopes. Contact our scientists to inquire about your HDX-MS project today.

Talk to Our Scientists.
We Have Sequenced 10,000+ Antibodies and We Are Eager to Help You.
Through next generation protein sequencing, Rapid Novor enables reliable discovery and development of novel reagents, diagnostics, and therapeutics. Thanks to our Next Generation Protein Sequencing and antibody discovery services, researchers have furthered thousands of projects, patented antibody therapeutics, and developed the first recombinant polyclonal antibody diagnostics.
Talk to Our Scientists.
We Have Sequenced 9000+ Antibodies and We Are Eager to Help You.
Through next generation protein sequencing, Rapid Novor enables timely and reliable discovery and development of novel reagents, diagnostics, and therapeutics. Thanks to our Next Generation Protein Sequencing and antibody discovery services, researchers have furthered thousands of projects, patented antibody therapeutics, and ran the first recombinant polyclonal antibody diagnostics