
Written by:
Yuning Wang, PhD
Updated: January 26, 2023
(Published: June 3, 2022)
Introduction
Structural information provides a great deal of understanding of how a protein works, which can allow us to elucidate molecular mechanisms underlying human diseases. Rapidly developing structural determination and analysis tools further enable researchers to tackle complicated proteins like membrane proteins, antibodies, and multiprotein complexes, providing insights into modern clinical drug design. This article will introduce the four levels of protein structure and the different techniques used for studying each structural level.
The Four Levels of Protein Structure
Primary Structure of Proteins
Proteins are made up of amino acids linked together by peptide bonds in different orders and different lengths, ranging from 30 amino acids to more than 100,000. Therefore, the structure of a protein begins with its amino acid sequence, which is considered its primary structure. Each type of protein possesses a distinct primary structure, which drives the bonding and folding of the linear amino acid chain, and ultimately determines the protein’s unique three-dimensional shape.
Secondary Structure of Proteins
The secondary structure of a protein refers to the local folding patterns on the polypeptide chain formed by intramolecular interactions between atoms of the backbone. The formation of the secondary structure is mainly driven by hydrogen bonding between amino groups and carboxyl groups on the polypeptide chain.
The most common types of secondary structure are α helix and β sheet. An α helix is made when the polypeptide chain turns around itself, forming a structural motif that resembles a spiral staircase. A β sheet is generated when multiple segments of a polypeptide chain lie side by side, creating a sheet-like structure held together by hydrogen bonds. Most proteins contain α helices and β sheets. For example, α helices are especially abundant in membrane proteins and hair cells (i.e., α-keratin), and β sheets are the main component of amyloid fibers in both animals and bacteria.
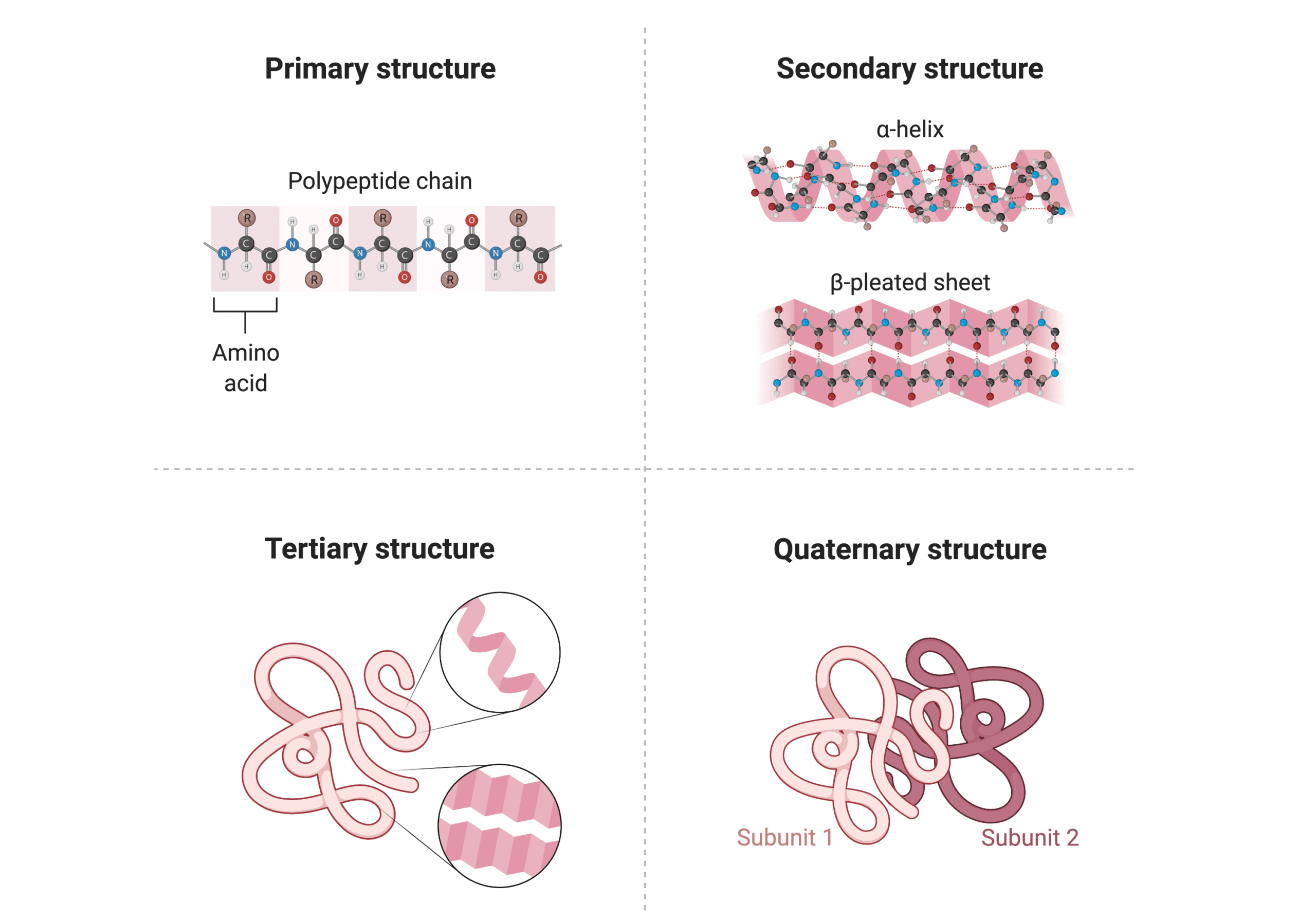
Tertiary Structure of Proteins
The overall three-dimensional conformation of a single polypeptide chain (a protein molecule) is referred to as the tertiary structure, which typically includes different elements of secondary structures such as α helices, β sheets, random coils, and loops. Bonds between side chains (R groups) of amino acids—including hydrophobic interactions, hydrogen bonds, and ionic bonds —contribute to the tertiary structure.
In addition, there is one type of covalent bond that can also contribute to tertiary structure: the disulfide bond. Disulfide bonds are a type of post-translational modification (PTM) formed between sulfur-containing side chains of cysteine residues, allowing distant parts of the protein to be held together. They are abundantly found in secretory proteins and extracellular domains of membrane proteins.
Quaternary Structure of Proteins
Many proteins are made up of more than one polypeptide chain to perform their function. The complete structure of such a protein is designated its quaternary structure, and each polypeptide chain is referred to as a subunit. The quaternary structure is stabilized by the same bonds as for the tertiary structure, including different noncovalent bonds and disulfide bonds. These bonds hold the subunits together and arrange themselves to form a larger protein complex.
How are Protein Structures Studied?
Various analytical, biophysical, and computational tools are currently available to study the four levels of protein structure, as highlighted in Table 1 below. In this section, we will discuss some of the widely adopted techniques for protein structural analysis.Various analytical, biophysical, and computational tools are currently available to study the four levels of protein structure, as highlighted in Table 1 below. In this section, we will discuss some of the widely adopted techniques for protein structural analysis.
Table 1. Current methods for the study of the four levels of protein structure.

Techniques for Studying Primary Structure
Structural analysis of a protein usually starts with the determination of its primary structure: the amino acid sequence. This can rely on two types of methods: 1) mass spectrometry (MS)-based protein sequencing, and 2) Edman degradation. The former is particularly useful to generate full sequences of large proteins (more than 40 amino acid residues). Nowadays, the advancement of MS-based proteomics has led to the advent of de novo protein sequencing, allowing for rapid and accurate sequence determination of any given protein. The primary structure of a protein can also be analyzed by means of peptide mapping, another MS-based method well suited for verifying sequence and identifying sequence variations like point mutations.
Techniques for Studying Secondary Structure
To study a protein’s secondary structure, the most common method is circular dichroism spectroscopy (CD). It measures different secondary structural elements in a protein, including α helix and β sheet, and random coil, based on their characteristic spectra in the far-UV region of the spectrum. These spectra can then be used to quantify the fraction of each secondary structural element in the protein. Another biophysical method, Fourier transform infrared (FTIR) spectroscopy, can also be used to estimate secondary-structural components in a protein by measuring the wavelength and intensity of infrared radiation (IR) absorption by a protein sample.
Techniques for Studying Tertiary Structure
The tertiary structure, or the three-dimensional structure, of a protein, is mainly determined using X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, or cryogenic electron microscopy (cryo-EM). Each method has its advantages and disadvantages. Generally, with these methods, different types of experimental data (i.e., X-ray diffraction, chemical shift) are gathered to measure distances between atoms and yield a three-dimensional model of the protein structure at the atomic level. To date, the 3D structures of more than 190,000 different proteins have been determined. Lastly, hydrogen-deuterium exchange (HDX)-MS is a powerful analytical technique often combined with other structural techniques to obtain important information on protein conformation, dynamics, and folding, therefore a more comprehensive picture.
Techniques for Studying Quaternary Structure
Finally, the quaternary structure of a protein can be studied using the same methods for the tertiary structure. In particular, the advancement of cryo-EM is gaining popularity for high-resolution visualization of large protein complexes with complicated quaternary structures. HDX-MS is also particularly useful for studying protein interactions, such as antibody-antigen binding.
Protein Structure Analysis with Bioinformatics
In addition to the analytical and biophysical methods mentioned above, a number of in silico tools have been rapidly emerging in recent years for the analysis of primary amino acid sequences and the prediction of secondary and tertiary protein structures. These computational tools can be leveraged for simple bioinformatic tasks, such as multiple sequence alignment and analysis (ie. Clustal Omega, MAFFT, MUSCLE), to more complicated tasks, such as secondary structure prediction (ie. JPred4, RaptorX, PEP2D) and the analysis of the protein’s 3D structure (ie. AlphaFold).
Protein Structural Analysis at Rapid Novor
Rapid Novor provides multiple services that can facilitate the analysis and characterization of protein structures.
- De novo protein sequencing – With our REmAb® protein sequencing service, obtain the protein’s primary structure by deriving its amino acid sequence using our proprietary MS-based protein sequencing and AI-assisted technology
- Peptide mapping – With our MATCHmAb™ peptide mapping service, confirm the protein’s primary structure and identify any potential anomalies that may influence the protein structure and/or function.
- HDX-MS epitope mapping – With our HDX-MS epitope mapping service, obtain the protein’s tertiary or quaternary structure by gaining insights into the conformation, dynamics, and folding of the protein or protein complex.
Please visit our services page to learn more about these technologies, and reach out to our scientists for inquiries.

Talk to Our Scientists.
We Have Sequenced 9000+ Antibodies and We Are Eager to Help You.
Through next generation protein sequencing, Rapid Novor enables reliable discovery and development of novel reagents, diagnostics, and therapeutics. Thanks to our Next Generation Protein Sequencing and antibody discovery services, researchers have furthered thousands of projects, patented antibody therapeutics, and developed the first recombinant polyclonal antibody diagnostics.
Talk to Our Scientists.
We Have Sequenced 9000+ Antibodies and We Are Eager to Help You.
Through next generation protein sequencing, Rapid Novor enables timely and reliable discovery and development of novel reagents, diagnostics, and therapeutics. Thanks to our Next Generation Protein Sequencing and antibody discovery services, researchers have furthered thousands of projects, patented antibody therapeutics, and ran the first recombinant polyclonal antibody diagnostics