 Written by:
Written by:
Bin Ma, PhD
Updated: January 26, 2023
(Published: February 3, 2017)
Which Techniques are used in Protein Sequence Determination and Analysis?
One of the most important pieces of information researchers need to know during early-stage antibody drug research and development is the sequence information of the antibody protein. With the advancement of mass spectrometry instrumentation and technologies, it is helpful, and sometimes critical, to conduct sequence analysis using mass spectrometry experiments. This article highlights three different technologies often used in this context:
- Intact Mass Analysis
- Peptide Mapping
- De Novo Protein Sequencing
Both of the first two methods, intact mass analysis and peptide mapping, require prior knowledge of an antibody’s amino acid sequence, but the third, de novo protein sequencing, does not. Instead, the amino acid sequence is directly derived from the LC-MS/MS data.
How Mass Spectrometry is used in Intact Mass Analysis
Intact mass analysis is the measurement and determination of the molecular weight of the intact antibody protein. It may measure the antibody protein in its native form, or it may measure the heavy and light chains separately after reduction. In the case of antibody fragments, it may also measure the fragments, such as Fab or VH+CH1.
When the antibody’s primary sequence is known, intact mass can provide extra information to confirm the primary sequence of the antibody protein. However, certain sequence variations, such as the Leucine/Isoleucine mutation and the swap of two amino acids, cannot be validated this way. More importantly, intact mass analysis can help derive the relative ratios of expressed glycoforms conferring biological activity or resulting in immunogenicity.
The general steps of a typical intact mass analysis are as follows:
- LC-MS on the purified antibody protein or its fragments to generate a full MS spectrum
- Deconvolution of the high-charged full MS spectrum
- Data analysis and interpretation
The image below illustrates a highly charged envelope of a target protein (B) as well as the corresponding deconvoluted spectrum (C).
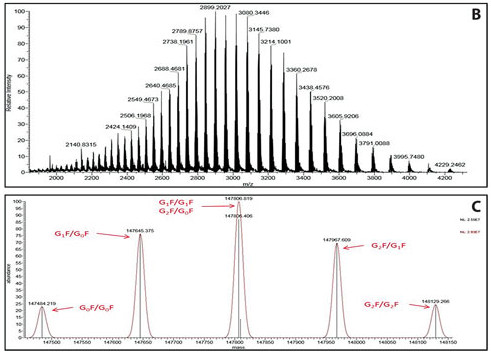
(Image credit: Genetic Engineering & Biotechnology News)
How Mass Spectrometry is used in Peptide Mapping
Peptide mapping is the analysis of peptides generated from the digestion of a protein by mass spectrometry. It is a comparative procedure and can be used as an identity test for proteins. It requires a reference, either a reference sequence, reference standard, or reference material, to compare and contrast with the target of interest.
Peptide mapping is particularly useful to find differences between two or more samples or conditions when visualized in the liquid chromatogram. It can also be used to confirm the sequence of the target protein against a known sequence and discover point mutations. The technology utilizes more information than intact mass measurement and therefore is more reliable. However, it still has difficulty detecting some subtle mutations such as the swap of two amino acids.
The general steps of peptide mapping are as follows:
- Digestion of the monoclonal antibody protein into peptides using one or more enzymes with diverse cleavage sites
- Separation of the peptides using chromatographic technologies such as reverse-phase HPLC
- Identification of the peptides (when MS/MS is used for the peptide identification, a database search engine such as Mascot and SEQUEST may be used)
- Reporting and visualization of the analysis by mapping the peptides to the LC Chromatogram or the known protein sequence
The image below illustrates a tryptic peptide mapping comparing three monoclonal antibodies.
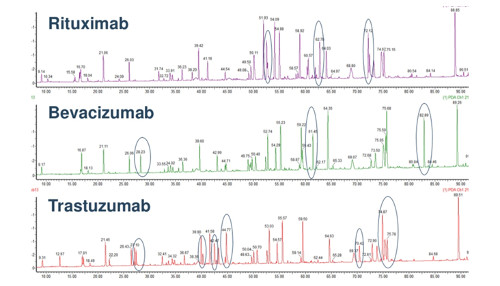
(Image credit: USP Biotherapeutics)
The image below shows how mapping peptides to a known protein sequence can help confirm the sequence information.
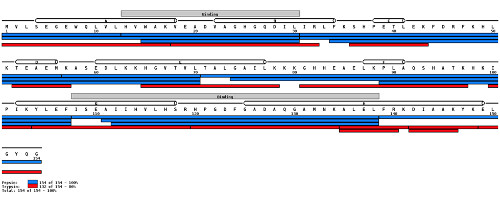
(Image credit: MS Tools)
How Mass Spectrometry is used in De Novo Protein Sequencing
De novo protein sequencing is the process of deriving the antibody protein sequence directly from the mass spectrometry data without prior knowledge of the sequence. It measures the peptides using high-mass accuracy instruments and derives the protein sequence based on the consensus of overlapping peptides as well as fragment ion information.
At the core of this technology is a de novo peptide sequencing algorithm that can produce both accurate de novo peptides and accurate scoring for each amino acid. This is particularly important for the complementarity determining region (CDR) of an antibody. The six CDR regions from the heavy and light chains are highly variable. This drives the diversity the immune system requires, but at the same time makes all the database sequences for those regions untrustworthy.
The general steps of de novo protein sequencing are as follows:
- Digestion of the monoclonal antibody protein to peptides using one or multiple enzymes with diverse cleavage sites
- Separation of the peptides using chromatographic technologies such as reverse-phase HPLC
- Producing tandem mass spectrum using high mass accuracy mass spectrometers
- De novo peptide sequencing to generate de novo peptides
- Assembly of de novo peptides into the full antibody protein sequence
The image below visualizes the peptides identified in a de novo sequenced protein by Rapid Novor’s antibody sequencing software. The bold fonts highlight the amino acids confidently supported by the MS/MS fragment ion peaks. The overlapping peptides collectively prove the correctness of each amino acid of the antibody protein.

(Image credit: Rapid Novor’s antibody sequencing software)
Conclusions
Each of the aforementioned technologies have their own unique advantages. The complexity of the experiments and data analysis increase significantly from intact mass analysis to de novo sequencing. As such, the required expertise and the associated cost, increase as well. If one is confident about the primary sequence and the purpose is only to confirm the original sequence against sequence variations which could significantly alter the intact mass of the whole protein, or against certain peptide’s mass or retention time, then intact mass analysis and peptide mapping, respectively, are the methods of choice due to lower cost and sometimes faster turnaround. If one requires the confidence of each amino acid of the antibody, then de novo protein sequencing is essential.
To learn more about Rapid Novor’s de novo antibody sequencing service, contact us!

Talk to Our Scientists.
We Have Sequenced 10,000+ Antibodies and We Are Eager to Help You.
Through next generation protein sequencing, Rapid Novor enables reliable discovery and development of novel reagents, diagnostics, and therapeutics. Thanks to our Next Generation Protein Sequencing and antibody discovery services, researchers have furthered thousands of projects, patented antibody therapeutics, and developed the first recombinant polyclonal antibody diagnostics.
Talk to Our Scientists.
We Have Sequenced 9000+ Antibodies and We Are Eager to Help You.
Through next generation protein sequencing, Rapid Novor enables timely and reliable discovery and development of novel reagents, diagnostics, and therapeutics. Thanks to our Next Generation Protein Sequencing and antibody discovery services, researchers have furthered thousands of projects, patented antibody therapeutics, and ran the first recombinant polyclonal antibody diagnostics