Written by:
 María Gerpe, PhD., and
María Gerpe, PhD., and
 Yuning Wang, PhD
Yuning Wang, PhD
July 19, 2021
Introduction
Proteins are composed of peptide chains, which in turn are made up of a string or linear sequence of amino acids (Figure 1A). Every amino acid has a basic structure containing an amino (-NH2) group and a carboxylic (-COOH) group (Figure 1B). To form a peptide, amino acids link to each other via a peptide bond, which involves the reaction between the carboxylic group of one amino acid and the amine group of another amino acid (Figure 1B). As such, the primary structure of a protein is typically recorded starting at the amino-terminal (N) end and continuing to the carboxyl-terminal (C) end. The primary protein structure may be directly sequenced from a sample of the protein itself or inferred from the DNA sequence.
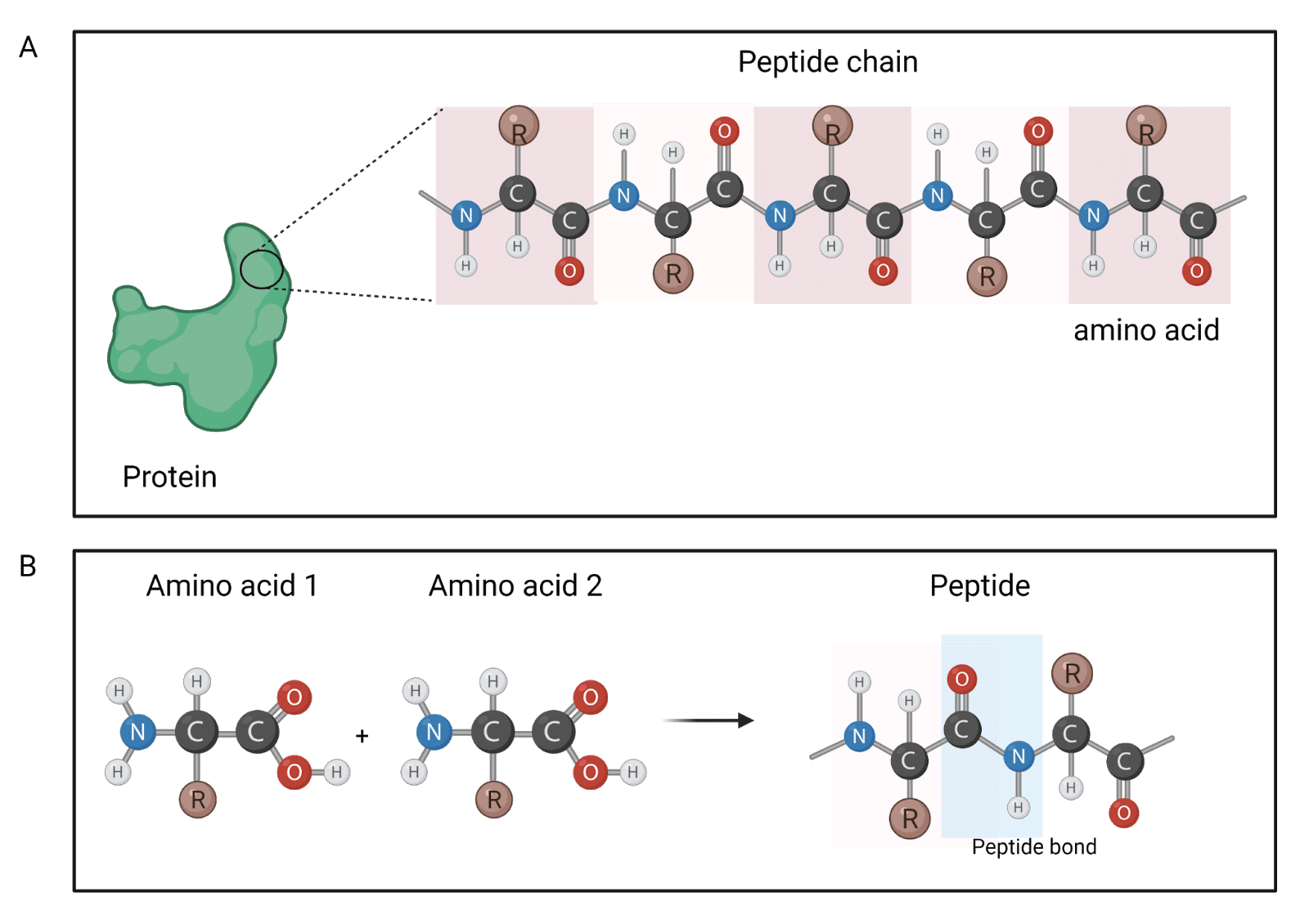
Figure 1. Illustration of protein building blocks. A protein is composed of peptide chains, each of which is made by a string of amino acids (A). Amino acids have an R group (carbon chain), and two functional groups: an amino group, and a carboxylic group. When two amino acids join together through a reaction between their carboxyl and amino groups, they form a peptide bond.
The amino acid sequence of a protein can provide insight into its three-dimensional structure, cellular location, function, and evolution. Many of these insights are discovered via the search for similarities with sequences which are already known and stored in online databases. Comparing a newly obtained protein sequence with the large bank of stored sequences can show similarities and relationships to help characterize proteins. Recent advances in machine learning and bioinformatics are also starting to allow predictions about the further developability and optimization of proteins (e.g., antibodies) for use as therapeutics or diagnostics.
Amino acid sequencing offers important information for the full understanding of a protein or peptide and the ability to identify a protein in a sample and classify its post-translational modifications. The process of ascertaining the amino acid sequence is known as amino acid sequencing.
De Novo Amino Acid Sequencing
De novo amino acid sequencing is synonymous with de novo antibody protein sequencing. At Rapid Novor, we specialize in de novo antibody protein sequencing to ascertain the amino acid sequence of an unknown protein by employing mass spectrometry and machine learning algorithms. Unlike other protein mass spectrometry methodologies, de novo antibody protein sequencing requires no access to the cell line and no prior knowledge of the nucleotide sequence. De novo amino acid sequencing has been able to expand on results beyond existing DNA databases. A recent example was the confirmation of discovery of Denisovian remains, when DNA information was scant. To assign amino acids, it uses Big Data-driven algorithms to fully extract all information from mass spectrometry data including peptide fragment masses from mass-to-charge ratios of tandem spectra.
Protein Sequencing Versus Peptide Sequencing
De novo protein sequencing and peptide sequencing are often used interchangeably, but they differ in terms of the level of analysis and the scope of information they provide.
Peptides, being smaller than proteins, can present challenges such as lower success rates for smaller or highly modified peptides. Additionally, peptides with ambiguous or overlapping fragment ions can complicate the identification process. Since there is typically not as much sequence overlap as in protein sequencing, the overall sequence confidence can be reduced.
Peptide sequencing is often a step in protein sequencing, providing sequence information for smaller fragments of the protein. However, due to the larger size of proteins, enzymatic digestion and fragmentation produce multiple peptide overlaps, offering high-confidence sequence information and a more complete understanding of the protein’s structure.
Sequencing Methodology
Each step of the de novo amino acid sequencing method requires expertise and experience in order to get the best results quickly (Figure 2).
- Sample preparation. Ensure the sample purity by first running SDS-PAGE. More specific and purpose built purification methods will get the best downstream results.
- Digestion. Use pepsin, trypsin or other enzymes to ‘cut’ the protein into peptides for analysis. Pay attention to the quality of the enzyme used for digestion. Also consider using multiple enzymes.
- LC-MS/MS. HPLC separates peptides, which are then fed into a mass spectrometer for analysis. It is essential to use a mass spectrometer built for proteomics. Care must be taken in the fragmentation method for the MS2, and in the selection window to ensure optimal coverage.
- Peptide de novo sequencing. Interpret each mass spectrum to determine the sequence of each peptide. Software is available for this purpose, but the latest, most advanced algorithms get answers faster with less ambiguity. Expert human interpretation may still be required post software analysis.
- Sequence assembly. Construct the full length protein sequence from the peptide sequences. Look for as much overlap as possible. Expert human interpretation may still be required post software analysis.
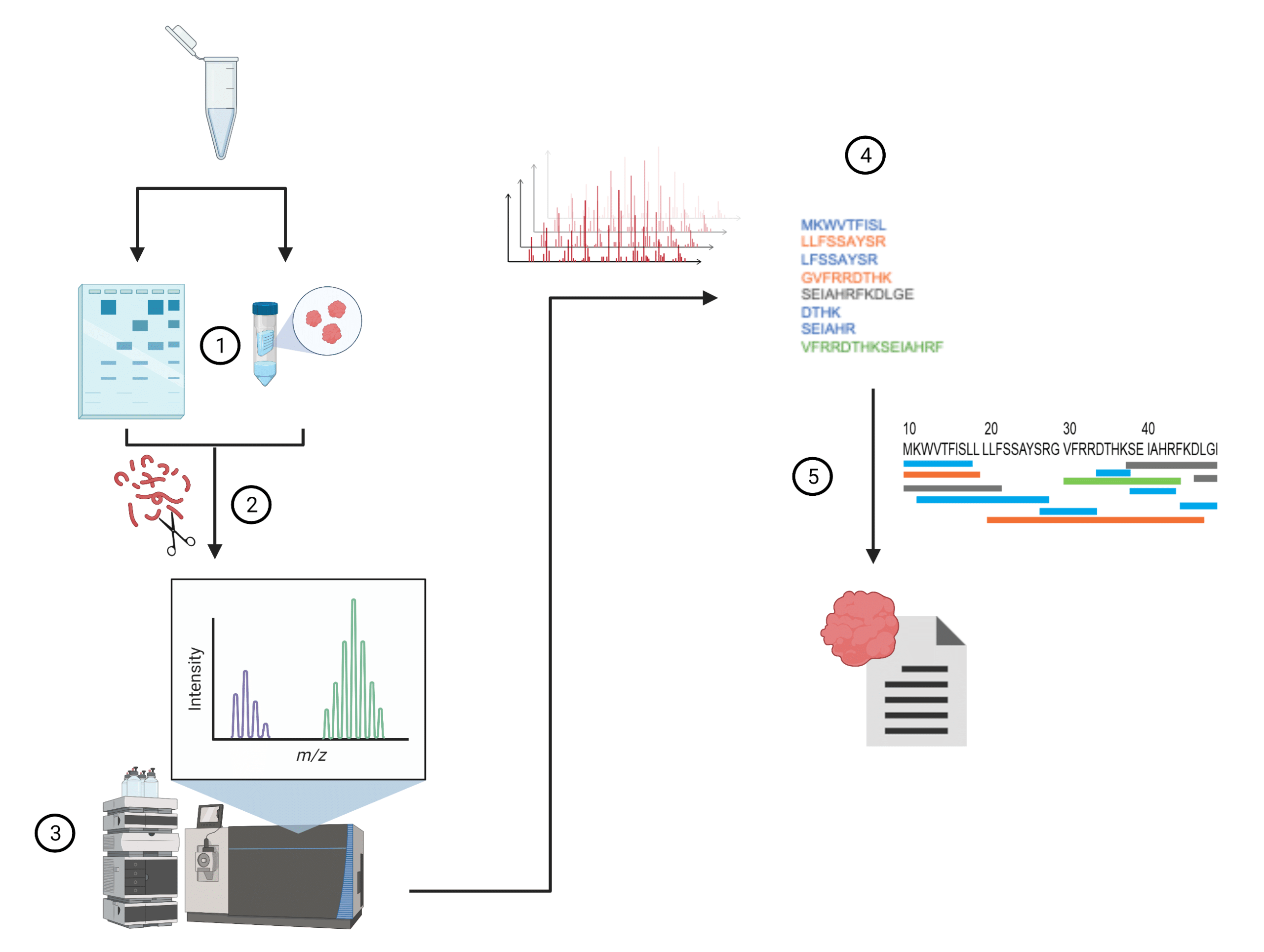
Figure 2. Infographic providing an overview of the methodology behind de novo amino acid sequencing.
While ESI-based MS instruments initially contained one mass analyzer and one ion detector, nowadays, ion cells are typically flanked by the ion source and the ion sensor and sandwiched between electrodes that can modulate the frequency and voltage to ‘select’ a window for the desired m/z ratio and then redirect the flow of ions for additional fragmentation, for instance.
The ion source, and mass analyzers and ion detectors are all kept under vacuum. An example of an ion cell that can act as either or both a mass analyzer and an ion detector is the ion trap.
As peptides travel between mass analyzers and ion detectors, collisions can fragment these peptides further via high-energy collision dissociation (HCD), or electron-transfer high-energy collision dissociation (EThcD). All of the internal systems feed into the instrument control. Essentially, at any of these points, data can be detected such that the user may select a specific range of ions and record the spectra of these fragments for data analysis.
A mass spectrometer that comprises different sequential cells with mass analyzer and ion detector capabilities is referred to as a tandem mass spectrometer. Mass spectrometers used to only be able to house a mass analyzer and an ion detector. However, mass spectrometry technology has evolved to such a sophisticated level that current tandem mass spectrometers house many more components than original instruments, conveniently fitting in one corner of a laboratory.
Rapid Novor De Novo Amino Acid Sequencing Solutions
De novo amino acid sequencing offers complete coverage of the protein sequence from the N-terminus to C-terminus with complete confidence. Rapid Novor’s de novo amino acid sequencing from Rapid Novor employs the most advanced software technology of its kind, and mass spectrometry methods that afford confidence for precise protein amino acid sequencing. This means for example that recombinantly expressed antibodies work just as expected, the first time.
At Rapid Novor, we work with smaller sample input, and provide a faster turnaround, without the need for hybridoma or previous understanding of the antibody protein or DNA sequence information. With our REmAb® antibody sequencing service, results of the antibody sequence or other protein sequence directly from a protein sample can be provided within a week. REpAb® polyclonal antibody sequencing service can be applied to polyclonal antibodies, sequencing full-length Abs directly from serum or a purified protein mixture.
If you would like to find out more about how Rapid Novor can help with amino acid sequencing, contact us today for more information.

Talk to Our Scientists.
We Have Sequenced 10,000+ Antibodies and We Are Eager to Help You.
Through next generation protein sequencing, Rapid Novor enables reliable discovery and development of novel reagents, diagnostics, and therapeutics. Thanks to our Next Generation Protein Sequencing and antibody discovery services, researchers have furthered thousands of projects, patented antibody therapeutics, and developed the first recombinant polyclonal antibody diagnostics.
Talk to Our Scientists.
We Have Sequenced 9000+ Antibodies and We Are Eager to Help You.
Through next generation protein sequencing, Rapid Novor enables timely and reliable discovery and development of novel reagents, diagnostics, and therapeutics. Thanks to our Next Generation Protein Sequencing and antibody discovery services, researchers have furthered thousands of projects, patented antibody therapeutics, and ran the first recombinant polyclonal antibody diagnostics