
Written by Yuning Wang, PhD
May 13, 2022
What is HDX-MS?
HDX-MS is a mass spectrometry-based technique which involves protein deuteration to distinguish amide hydrogens participating in secondary structures. HDX-MS is used to examine protein-protein interactions, including antibody-antigen interactions, protein conformational changes, multi-protein complexes, and small molecule interactions with proteins.
Underlying Theory of HDX-MS
When a protein is dissolved in a solvent, amide hydrogens of the backbone undergo continual exchanges with hydrogens present in the solvent. As such, when using a deuterated solvent (i.e., D2O), the exchange process within the protein involves the substitution of amide hydrogen atoms by deuterium atoms (Figure 1).
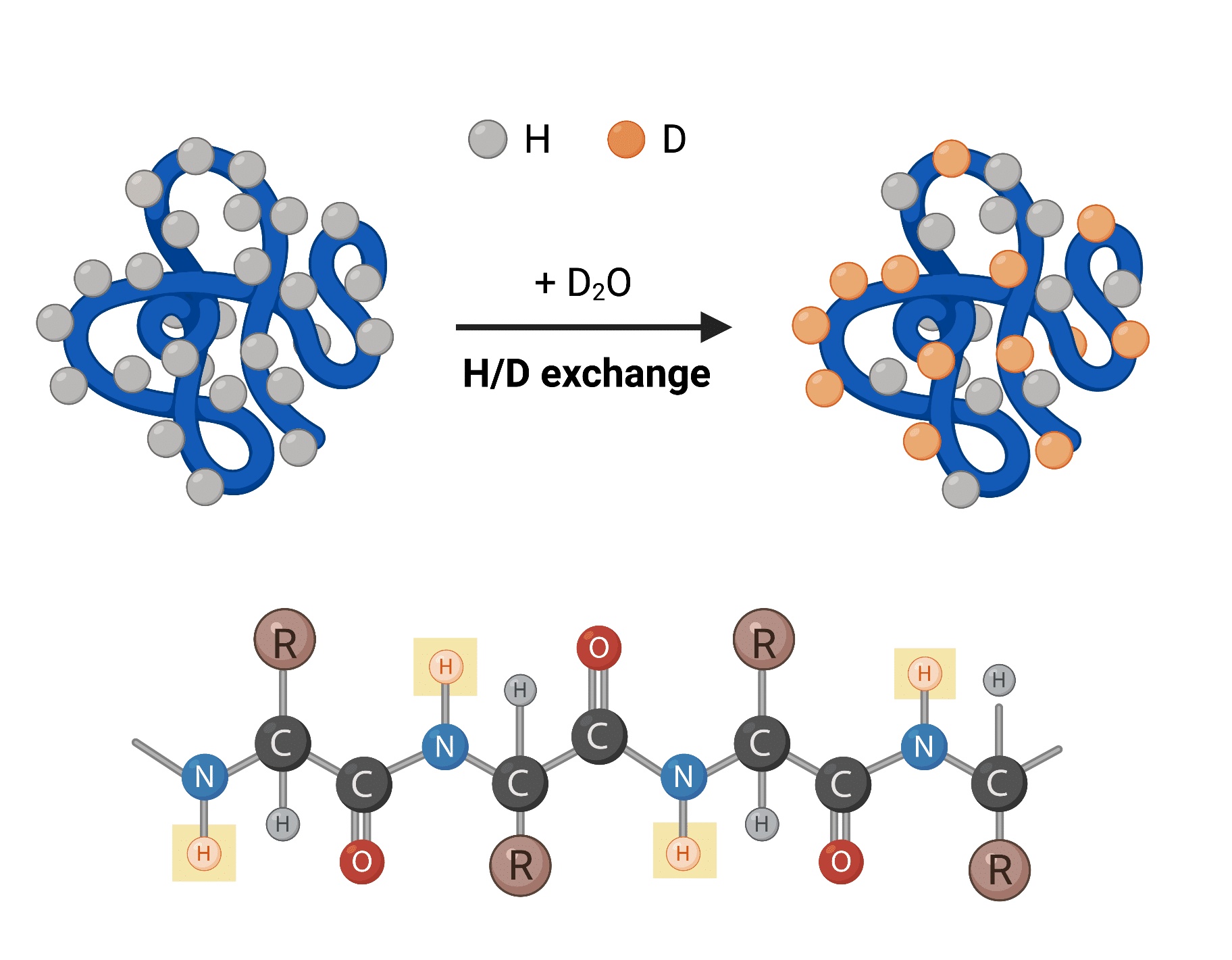
Figure 1. Top: hydrogen-deuterium (H/D) exchange process of protein dissolved in D2O. Bottom: protein chain with backbone amide hydrogens capable of undergoing H/D exchange highlighted in yellow boxes.
The rate of hydrogen-deuterium exchange is dependent on the solvent accessibility and folded state of the protein, which can be used to infer information on protein structure and dynamics. HDX is commonly measured by nuclear magnetic resonance (NMR) spectroscopy or mass spectrometry (MS); the latter is referred to as HDX-MS.
HDX-MS Workflow
There are two conventional workflows for HDX-MS experiments: the ‘top-down’ approach, where intact protein is introduced into the mass spectrometer, and the more commonly-used ‘bottom-up’ approach involving protein digestion.
The general workflow of the bottom-up HDX-MS method is as follows (Figure 2):
- Labeling – incubate different fractions of protein in a D2O buffer to allow for H/D exchange at different time points (10s to several hours)
- Quenching – reduce the pH and temperature to prevent further exchange or back-exchange
- Digestion – digest the protein with proteases into shorter peptides for analysis
- LC-MS – inject and analyze peptides with mass spectrometer resulting in a set of LC-MS chromatograms that correspond to the set of peptides at different labeling times
- Data analysis – determine the uptake/incorporation of deuterium within digested peptides using data analysis software and plot across multiple time points
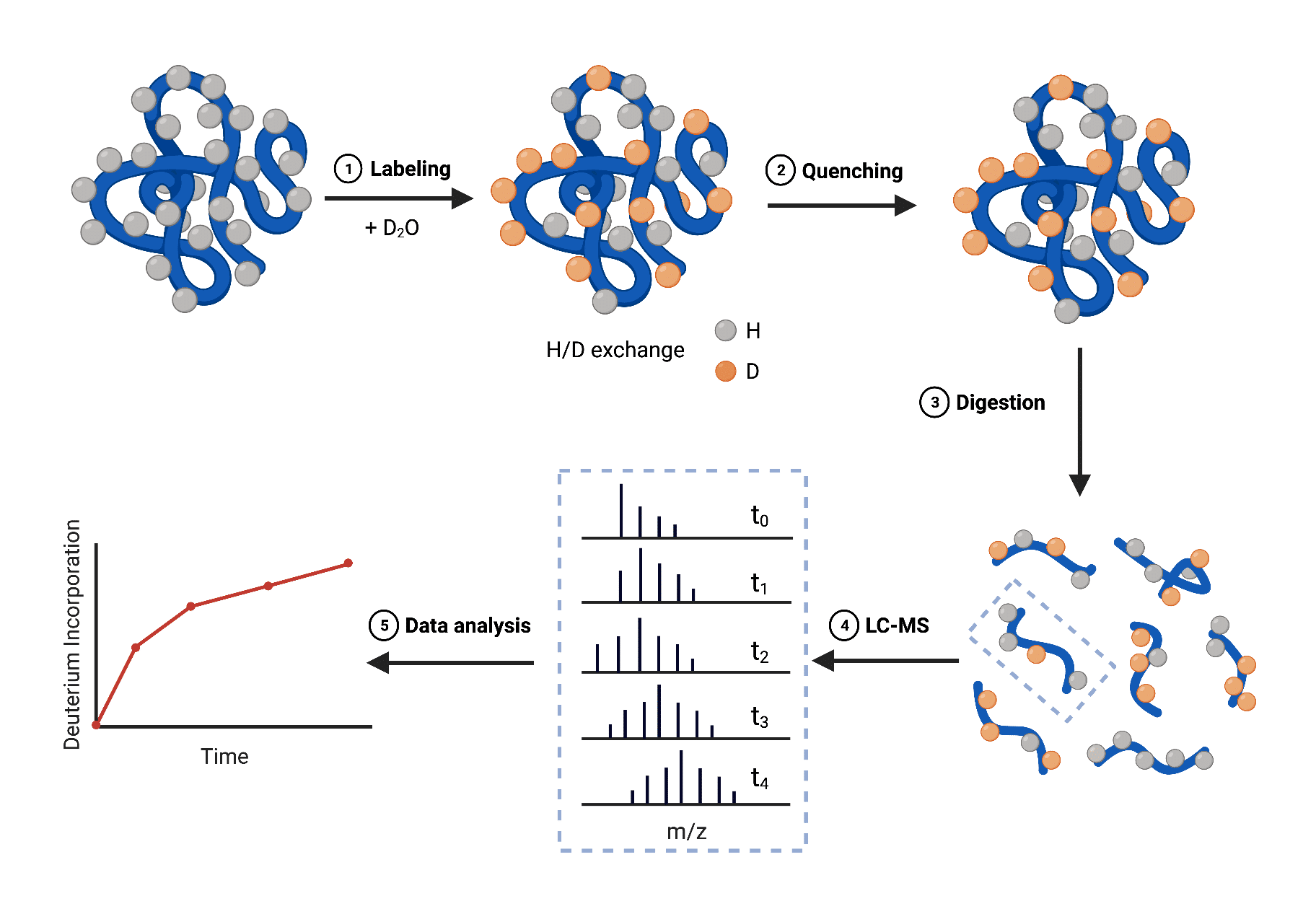
Figure 2. Schematic overview of HDX-MS workflow employing a bottom-up approach
The deuterium uptake information provides a site-specific HDX profile of the protein. Differences in HDX data of proteins in different states (e.g., folded and unfolded, bound and unbound) can be compared to detect local conformational changes. For instance, HDX-MS experiments can allow for the identification of the binding site in a protein-ligand complex by comparing HDX data of the bound and unbound state of the protein. HDX data are often mapped out on the protein’s three-dimensional structure to characterize structural and dynamic features.
Applications of HDX-MS
HDX-MS is a versatile technique with a number of applications (Figure 3), some of which are described below.
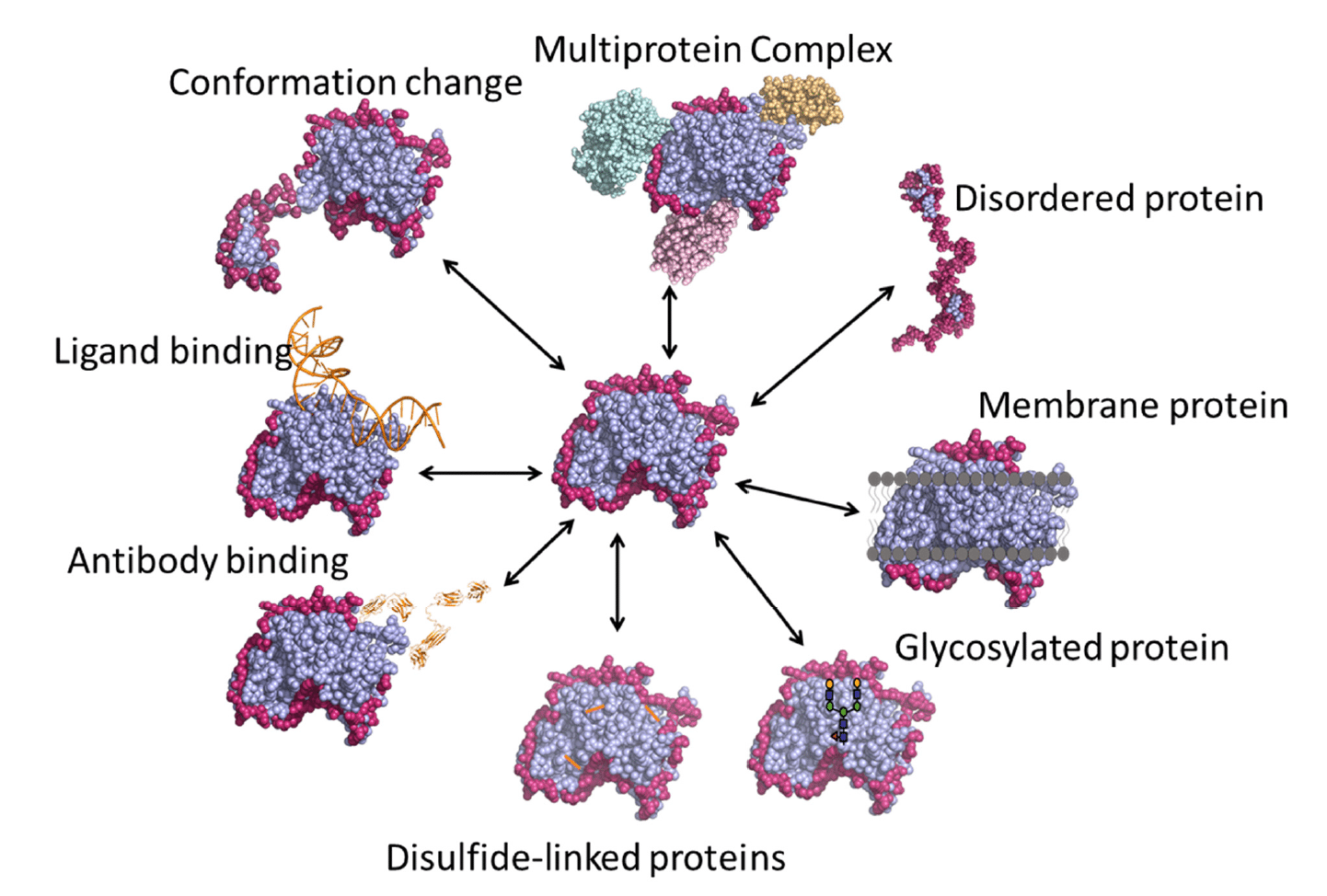
Figure 3. Various applications of HDX-MS. Image credit: https://www.mdpi.com/2227-9059/8/7/224
Characterizing Protein Conformation and Comparability
HDX-MS can be used to compare different conformers of a protein to identify conformational changes. In the study of genetic disorders, HDX data can help elucidate the effects of pathogenic mutations on the protein structure, folding, and stability, offering insights into the molecular mechanisms of the diseases and providing drug design possibilities. For example, a lot of research has been conducted by HDX-MS to elucidate protein misfolding and aggregation in neurodegenerative diseases, including Alzheimer’s disease and Parkinson’s disease.
Dissecting Protein-ligand Binding
HDX-MS is an indispensable tool to capture protein structural or dynamic changes induced by binding to ligands, including small molecules, peptides, nucleotides, or other proteins. The experiment typically measures HDX of the protein in both ligand-free (apo) and ligand-bound (holo) states. Differentiating the HDX profiles between the two states allows for the identification of the binding interface. Importantly, HDX-MS can reveal both orthosteric and allosteric effects on the protein molecule upon ligand binding and is sensitive enough to characterize weak binding.
Probing Chemical Modifications
Post-translational modifications (PTMs) play important roles in regulating protein functions and activities. In recent years, HDX-MS has been applied to study proteins encompassing PTMs, especially glycosylations and disulfide bonds. For example, researchers have used HDX data to characterize the glycosylation state of proteins and detect how glycosylations can impact the overall structure.
Additional applications of HDX-MS include studying conformational dynamics of membrane proteins and intrinsically disordered proteins, structural characterization of multiple-protein complexes, etc.
HDX-MS Advantages
Compared to conventional techniques of structural biology, such as X-ray crystallography and solution NMR, HDX-MS possesses multiple benefits.
- Low amount of sample required: HDX-MS requires as little as 0.1 mg of protein, which is far less than X-ray crystallography or NMR.
- No size limit: Unlike NMR, there is no theoretical size limit to the proteins being analyzed by HDX-MS, making it ideally suited for studying large biomolecular complexes like membrane proteins.
- No need for chemical labeling: HDX-MS does not need additional chemical labeling, such as isotopic labeling required for NMR.
- Broader applicability: HDX-MS can study a wider variety of proteins and binding properties, such as intrinsically disordered proteins and weakly-bound complexes, which often pose challenges to crystallography or NMR.
- High efficiency: HDX-MS experiments usually can be done within weeks, whereas structural determination by X-ray crystallography or NMR can take months.
HDX-MS Epitope Mapping of Antibodies
With a focus on antibodies, HDX-MS epitope mapping is the process of identifying the antibody binding site on the antigen surface. Epitope mapping follows the general workflow of HDX-MS, with the antigen and antigen-antibody complex being analyzed. Through epitope mapping, HDX-MS provides essential knowledge on antibody-antigen binding mechanisms. Such knowledge can help identify suitable antibodies as drug candidates for development, as well as evaluate immune responses induced by vaccines or other therapeutics.
Epitope mapping is a critical step in therapeutic antibody and vaccine development, especially in the lead generation stage of drug discovery. The FDA and EMEA both recommend epitope mapping as a tool for probing information about the specific binding sites of therapeutic and diagnostic antibodies.
Types of Antibody Epitopes
Typically, there are two types of antibody epitopes (Figure 4): 1) linear epitopes, where a segment of continuous amino acids of the antigen participate in antibody binding, and 2) conformational epitopes, composed of sequentially discontinuous amino acids that form a three-dimensional structure recognized by the antibody. The majority of human antibodies bind target antigens by conformational epitopes; therefore they can only recognize the corresponding antigens in native conformations.
HDX-MS epitope mapping is able to map both linear and conformational epitopes. It is considered to be the most effective method to rapidly supply near-complete information about epitope structure.
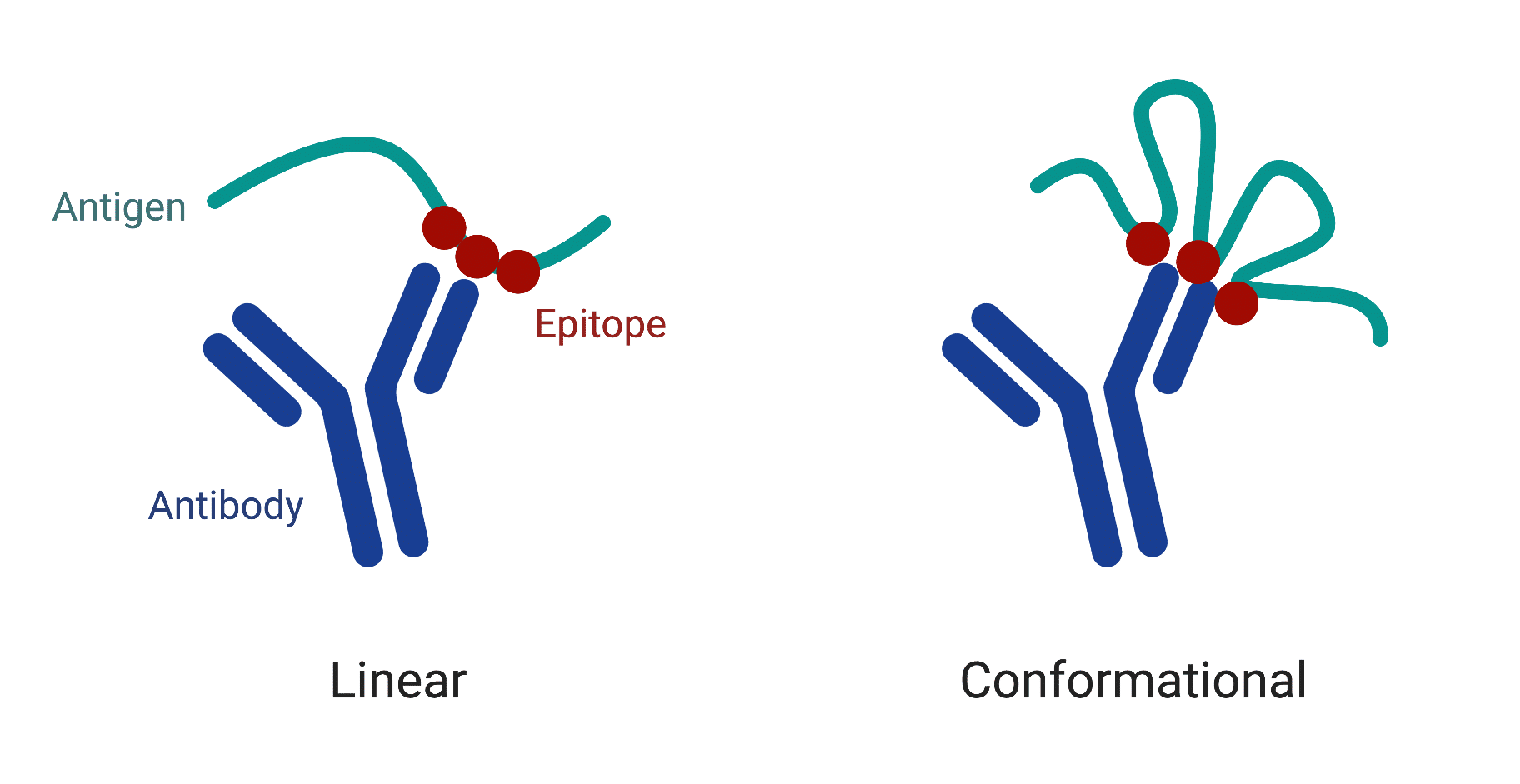
Figure 4. Two types of antibody epitopes
HDX-MS Epitope Mapping at Rapid Novor
HDX-MS epitope mapping service at Rapid Novor is a high-throughput, high-resolution method of choice for rapid binding site characterization in therapeutic and diagnostic antibody development. Built on over 20 years of innovation in proteomics and protein chemistry, we provide a bottom-up MS solution to rapidly and accurately determine both linear and conformational epitopes. Reach out to our scientists for inquiries about HDX-MS epitope mapping and/or other proteomics technology and services.

Talk to Our Scientists.
We Have Sequenced 10,000+ Antibodies and We Are Eager to Help You.
Through next generation protein sequencing, Rapid Novor enables reliable discovery and development of novel reagents, diagnostics, and therapeutics. Thanks to our Next Generation Protein Sequencing and antibody discovery services, researchers have furthered thousands of projects, patented antibody therapeutics, and developed the first recombinant polyclonal antibody diagnostics.
Talk to Our Scientists.
We Have Sequenced 9000+ Antibodies and We Are Eager to Help You.
Through next generation protein sequencing, Rapid Novor enables timely and reliable discovery and development of novel reagents, diagnostics, and therapeutics. Thanks to our Next Generation Protein Sequencing and antibody discovery services, researchers have furthered thousands of projects, patented antibody therapeutics, and ran the first recombinant polyclonal antibody diagnostics