 Written by Yuning Wang, PhD
Written by Yuning Wang, PhD
October 21, 2021
Updated on April 22, 2022
What is Peptide Mapping?
Peptide mapping is a widely used analytical technique to identify or verify a protein’s primary structure (amino acid sequence and chemical modifications). Briefly, peptide mapping identifies a protein by first determining peptides’ molecular weights and then matching these peptide masses to a protein’s calculated theoretical mass.
How is Peptide Mapping Performed?
Peptide mapping typically employs a bottom-up mass spectrometry approach. Scientists analyze peptides generated from digestion of an isolated protein, or a protein mixture. The basic workflow of peptide mapping involves enzymatic digestion of the protein followed by separation of the resulting peptides via liquid chromatography prior to mass spectrometry assessment. The procedure generates a ‘fingerprint’ or set of unique peptides of the interrogated protein. The masses/spectra of the unique peptides identified are compared to theoretical masses/spectra from sequences found in a proteomics database (Figure 1).
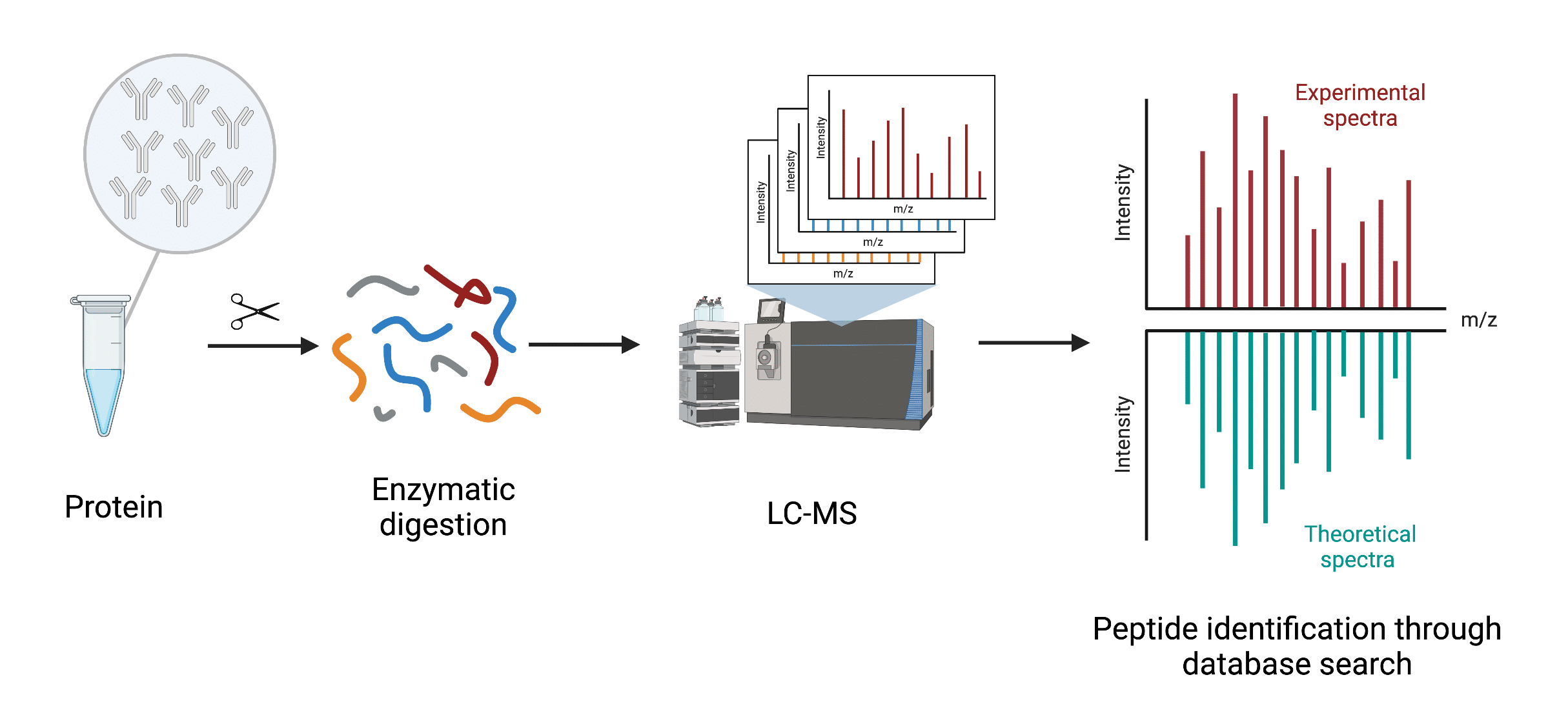
Figure 1. General workflow of peptide mapping utilizing bottom-up mass spectrometry-based proteomics
Unlike de novo protein sequencing, which resolves protein sequences down to the individual amino acid without a reference sequence, peptide mapping is dependent on sequence databases to identify a protein of interest.
Why is Peptide Mapping Needed?
As monoclonal antibody and recombinant protein drugs are becoming the fastest growing sectors of the pharmaceutical industry, careful inspection of the complete product properties (e.g., purity, biochemical properties, and stability) is critical throughout development and manufacturing. This is especially the case with antibodies which are often heterogeneous in nature.
Furthermore, antibodies’ various post-translational modifications (PTMs) (Figure 2) may occur over the molecule’s lifecycle. PTMs play significant contributions to antigen-binding, folding, and biological function of antibodies, consequently and directly impacting drug efficacy, stability, and safety. For example, glycosylation, one of the most common PTMs in antibodies, plays an important role in regulating many drug side effects including antibody-dependent cellular cytotoxicity and phagocytosis.
In addition to PTMs, other artifacts (e.g., aggregation, degradation, and contamination) which contribute to antibody heterogeneity can also be introduced during manufacturing and long-term storage. The latter may not only gravely impact manufacturing, but may also result in severe immunogenicity during antibody drug administration to patients.
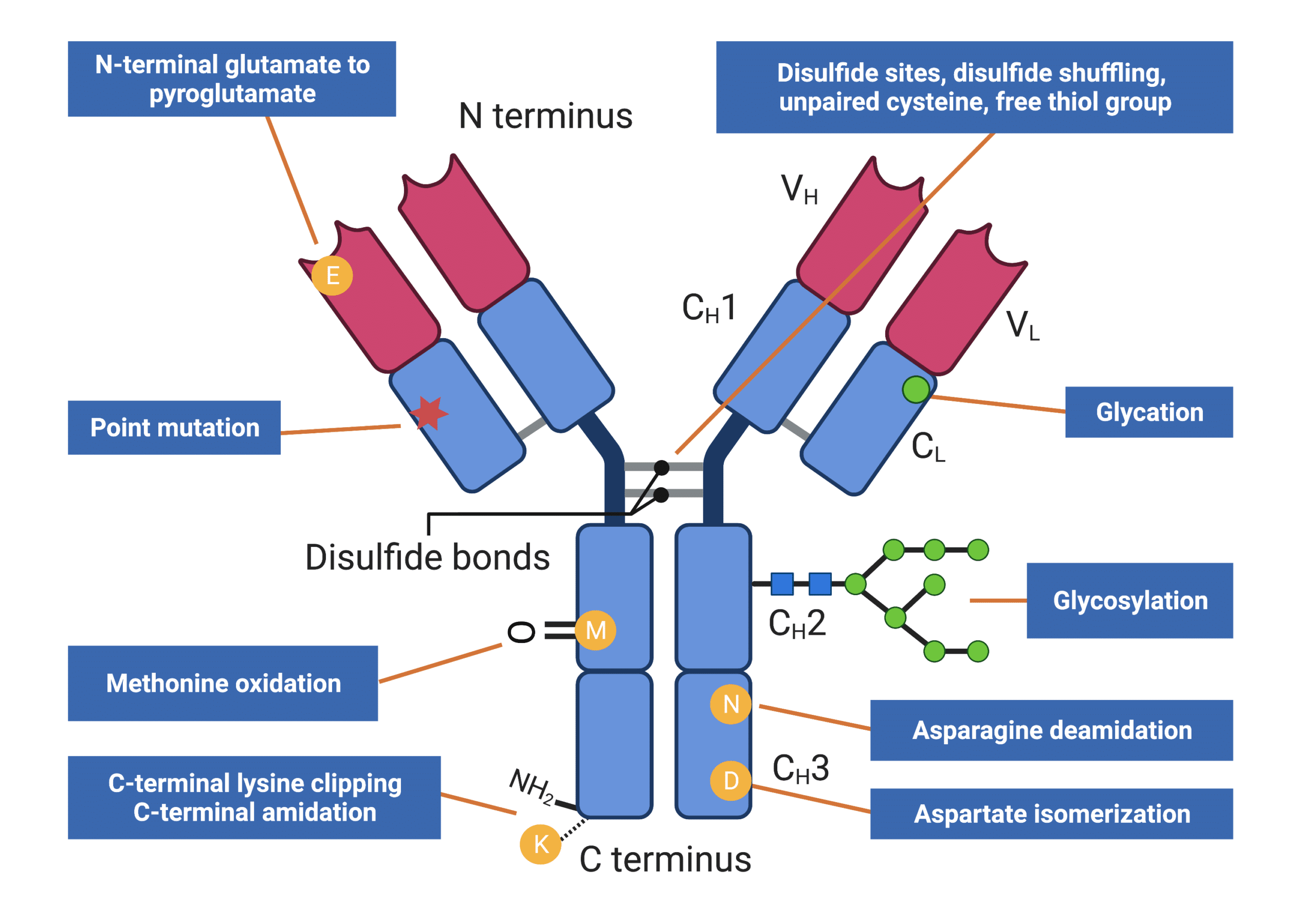
Figure 2. Infographics showing types of antibody heterogeneity that can be analyzed by peptide mapping
Applications of Peptide Mapping
Peptide mapping is widely applied in the biopharmaceutical industry throughout the development of therapeutic proteins, such as monoclonal antibodies and antibody-drug conjugates (ADCs). It is used for primary structural identification and product comparability testing. Peptide mapping is also an essential step in manufacturing process monitoring and quality assurance/quality control (QA/QC) to ensure no undesired change has occurred to the product.
Specifically, peptide mapping can be used to note a range of properties of the target protein, which include the following,
- Verification of the primary structure (amino acid sequence) of antibodies and antibody-derived therapeutics.
- Identification of point mutations and sequence variants between two or more samples.
- Monitoring of post-translational modifications (PTMs) present on proteins (Figure 2) (e.g., glycosylation, asparagine deamidation, methionine oxidation, aspartate isomerization, C-terminal clipping, etc).
- Determining disulfide linkage locations, unpaired cysteines, free thiol groups, and detecting the occurrence of disulfide shuffling.
- Identification of sites of degradation in a therapeutic antibody as the molecule degrades upon storage.
- Examine contamination and other dominant forms in the protein sample
Impacts of not utilizing peptide mapping
Failing to identify a product’s heterogeneity through peptide mapping can impact reproducibility and hamper drug development. Recent studies have shown that at least 50% of reagent antibodies cannot recognize their targets specifically, and a half or more preclinical studies cannot be reproduced. The Food and Drug Administration (FDA) and European Medicines Agency (EMA) both recommend extensive evaluation and testing of such changes including protein sequences, higher-order structures, and PTMs. Having a robust workflow that includes routine monitoring and validation of the protein, including protein sequence confirmation, is vital to ensure antibody quality and reproducibility.
Limitations of Traditional Peptide Mapping
Traditional Peptide Mapping Suffers from Low Resolution
Since conventional peptide mapping does not employ tandem mass spectrometry and peptide fragment data analysis (as would be employed in de novo protein sequencing), it suffers from lower resolution. Foregoing assessment at the peptide level results in the inability to distinguish between isobaric or same-mass peptides or amino acid combinations.
As such, bias may be introduced when performing database searching; peptide X, which shares the same/similar mass as peptide Y, might be identified as the same despite one or more isobaric or isomeric combinations. Some amino acid combinations such as AlaAsn and GlyGln; SerGlu and ThrAsp, GlyGlu and SerVal, or Arg and GlyVal have similar mass and may confound traditional peptide mapping approaches.
Traditional Peptide Mapping is Lengthy and Costly
Because it depends on database searching, it may not be clear whether coverage is good enough to correctly identify the protein simply based on the mass spec experiment. If a significant portion of the protein sequence is not covered by the tandem mass spec results, the experiment may have to be repeated.
The latter may result in delays, and limit the utility of the technique as a routine sequence check. Using tandem mass spec instrumentation that would have higher resolution, together with purpose-built bioinformatics software that would allow coverage to be assessed in real-time (and hence allow the mass spectrometry settings to be adjusted in real time), presents an attractive solution that would minimize time and budget spent.
MATCHmAb™: Next Generation Proteomics for Rapid Peptide Mapping
At Rapid Novor, we have developed a fast and robust protein sequence validation service, MATCHmAb™. MATCHmAb™ was developed from our established de novo protein sequencing REmAb® technology for precise sequence verification (Figure 3). As a result, MATCHmAb™ is capable of overcoming various conventional peptide mapping challenges. In contrast to traditional peptide mapping, MATCHmAb™ is
- Faster
- High-throughput (able to confirm protein sequences in hours to days)
- Capable of examining a sequence at the amino acid level
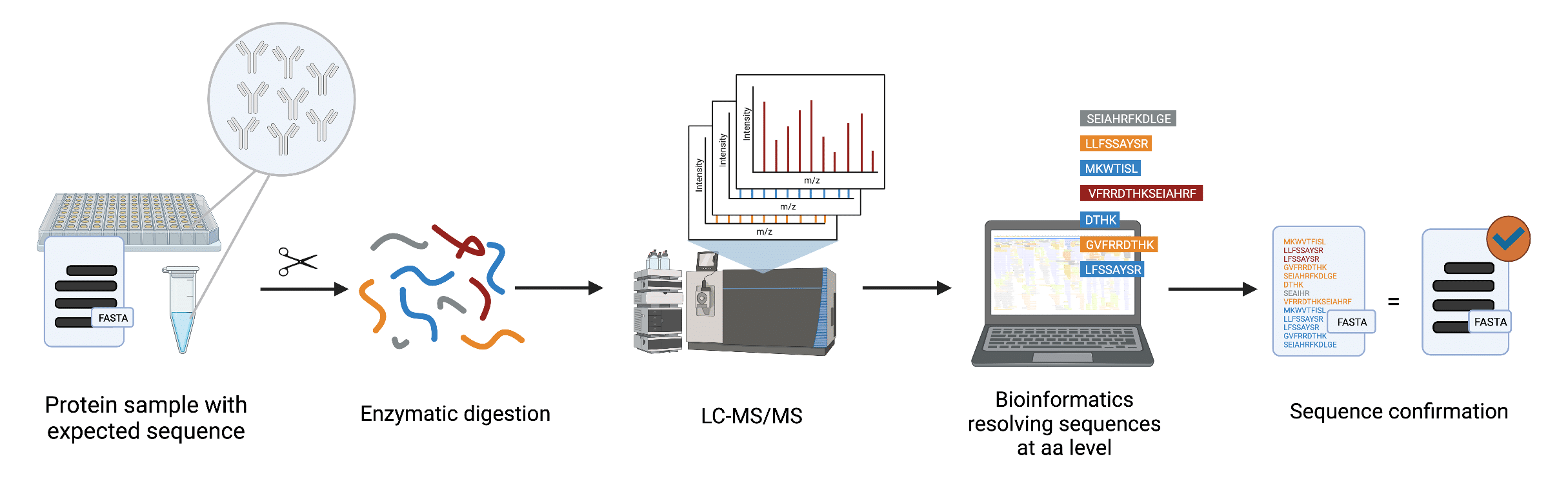
Figure 3. Workflow of MATCHmAb™ employing tandem mass spectrometry (LC-MS/MS) and our proprietary peptide identification software capable of resolving each amino acid of the peptide sequence
MATCHmAb™offers an easy check for long-lasting peace of mind. If you would like to find out more about how MATCHmAb™ can help with your project, contact us HERE.

Talk to Our Scientists.
We Have Sequenced 10,000+ Antibodies and We Are Eager to Help You.
Through next generation protein sequencing, Rapid Novor enables reliable discovery and development of novel reagents, diagnostics, and therapeutics. Thanks to our Next Generation Protein Sequencing and antibody discovery services, researchers have furthered thousands of projects, patented antibody therapeutics, and developed the first recombinant polyclonal antibody diagnostics.
Talk to Our Scientists.
We Have Sequenced 9000+ Antibodies and We Are Eager to Help You.
Through next generation protein sequencing, Rapid Novor enables timely and reliable discovery and development of novel reagents, diagnostics, and therapeutics. Thanks to our Next Generation Protein Sequencing and antibody discovery services, researchers have furthered thousands of projects, patented antibody therapeutics, and ran the first recombinant polyclonal antibody diagnostics