 Written by: Genya Gorshtein, MSc
Written by: Genya Gorshtein, MSc
Published: June 10, 2024
Introduction
Epitope mapping elucidates the antibody function by identifying specific binding sites on the antigen that interact with the antibody.
Alanine scanning mutagenesis is a commonly employed method for epitope mapping, involving the systematic substitution of individual amino acid residues to identify critical residues essential for antibody-antigen binding. Another method for epitope mapping utilizes hydrogen-deuterium exchange mass spectrometry (HDX-MS) to analyze the dynamic interaction between proteins by measuring the exchange rate of hydrogen with deuterium from the solvent.
In this discussion, we will compare the use of alanine scanning mutagenesis versus HDX-MS for epitope mapping of antigen-antibody interactions and consider important factors when selecting these methods.
Alanine Scanning Mutagenesis Epitope Mapping
Alanine scanning mutagenesis for epitope mapping involves substituting individual amino acids for alanine to identify the amino acid residues that play a role in antibody-antigen binding and other protein-protein interactions (Figure 1). Substituting with alanine removes all atoms past the β-carbon of the side chain, enabling the effects of individual alanine mutations to be used for identifying the roles of individual side chains.
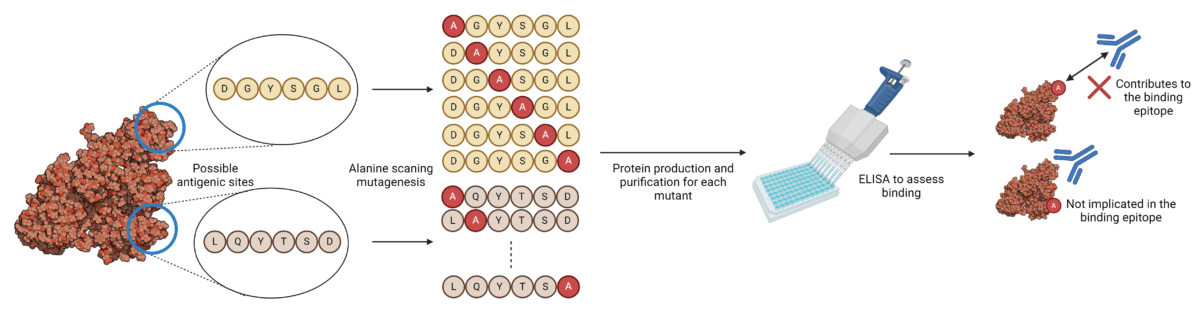
Figure 1. Alanine scanning mutagenesis workflow for epitope mapping of antibody-antigen interactions.
Mutant versions of the target antigen must be produced and purified, after which the binding is tested against antibodies for each mutant separately, to identify binding epitopes. Depending on the size and complexity of the target antigen, this could mean generating hundreds of different mutants. Residues whose mutation abrogates binding most severely are hypothesized to contribute to the binding epitope and paratope between the antibody and antigen.
Advantages of Alanine Scanning Mutagenesis
There are several advantages to alanine scanning mutagenesis for epitope mapping:
- Critical residue identification: Provides detailed information at the single amino acid level about the importance of specific residues in the binding interaction.
- Addresses energetics of binding interactions: By substituting individual amino acid residues with alanine, the changes in binding affinity can be measured between different mutants. This method enables researchers to identify and quantify the contributions of specific residues to the stability and strength of the protein-protein interaction.
- Accessible technology: Most laboratories and research facilities have in-house capabilities to perform site-directed mutagenesis and generate mutant plasmids for their proteins of interest. Furthermore, analysis of binding interactions is typically conducted using ELISAs or Western blots, which are highly accessible and cost-effective technologies that do not require complex or convoluted data analysis.
Disadvantages and Limitations of Alanine Scanning Mutagenesis
While alanine scanning can pinpoint the effects of antibody-antigen interactions down to a single amino acid, there are several disadvantages to this approach that must be addressed when deciding on methods to evaluate binding epitopes.
- Labor intensive and low-throughput: Producing and purifying multiple to hundreds of mutant proteins is time consuming, and resource-intensive. Additionally, this method is limited by the number of mutants that can be feasibly produced and analyzed.
- Limited mutation scope: Alanine is chosen because it is small and non-reactive, removing side-chain interactions without introducing new ones. However, this means that the method only reveals how the removal of side-chain atoms affects the interaction. It does not provide information on how replacing a residue with another amino acid, which could introduce different chemical properties (such as charge, hydrophobicity, or hydrogen bonding capacity), would impact the binding.
- Single residue analysis: Alanine scanning mutagenesis focuses on the impact of altering one residue at a time. This means it cannot reveal the combined or synergistic effects of mutating multiple residues simultaneously, possibly underestimating the effects of other residues belonging to the functional epitope.
- Conformational epitope mapping: Conformational epitopes are formed by residues that are brought together in the three-dimensional structure of the protein but may be distant in the primary sequence. Site-directed mutagenesis typically examines one residue at a time and may miss the broader context of how multiple residues contribute to the epitope.
To address the limitations of single residue analysis, a combinatorial mutagenesis scanning approach can be utilized, where several positions within a potential antigenic region are mutated to determine the impact of multiple residues on the binding epitope. However, mutations can sometimes cause local or global conformational changes that indirectly affect antibody binding. Distinguishing between direct impacts on the epitope and indirect structural perturbations can be challenging.
While alanine scanning has its advantages and is a commonly employed method for epitope mapping of antibody-antigen interactions, it should be combined with techniques that take into account the detailed three-dimensional structure of the antibody-antigen interaction to provide a more comprehensive understanding.
HDX-MS Epitope Mapping
HDX-MS is a mass spectrometry-based technique used to examine protein-protein interactions, including antibody-antigen interactions, protein conformational changes, multi-protein complexes, and small molecule interactions with proteins.
HDX-MS measures the exchange rate of amide hydrogen atoms with deuterium (D2O) when exposed to deuterium at different time points (Figure 2). By comparing the deuteration levels between bound and unbound states of the antibody-protein complex, the regions protected by antibody binding can be identified. These changes provide insights into which regions of the protein are protected (less exchange) and which are exposed (more exchange). Regions of the antigen involved in binding to the antibody show reduced deuterium uptake. The data can reveal conformational changes and dynamics of the antigen upon antibody binding, offering a deeper understanding of the interaction.
For a more detailed understanding of the theory of HDX-MS, visit our previous articles: What is HDX-MS Epitope Mapping and Protein Characterization by HDX-MS.
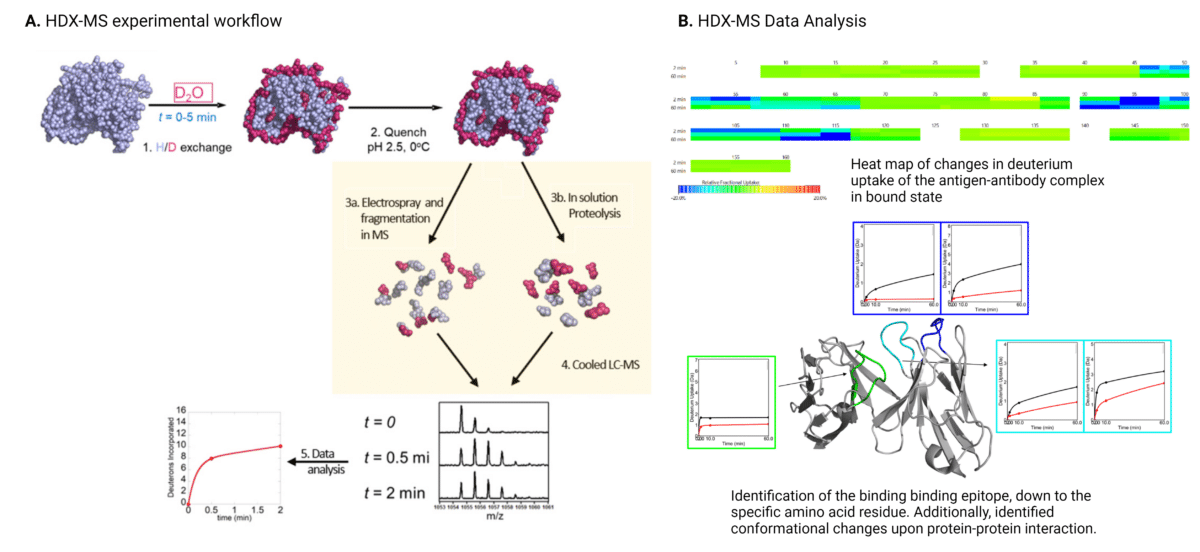
Figure 2. Workflow (A) and data analysis (B) for HDX-MS epitope mapping of antibody-antigen interactions. In the HDX-MS heat map depicting changes in deuterium uptake, green represents regions showing no change in deuterium uptake, cyan represents regions showing slight decrease in deuterium uptake, and blue represents regions showing strong decrease in deuterium uptake.
Advantages of HDX-MS Epitope Mapping
HDX-MS for epitope mapping has many advantages:
- Throughput: HDX-MS requires fewer sample preparation and purification steps compared to mutagenesis. Additionally, the exchange process and subsequent mass spectrometry analysis can be performed relatively quickly. HDX-MS systems are equipped with automated sample handling and data acquisition capabilities, which streamline the process and increase throughput.
- Not limited by antigen size or complexity: Unlike some other structural methods like X-ray crystallography or Cryo-EM, HDX-MS can be applied to proteins of various sizes and complexities, including large multi-subunit complexes and membrane proteins. This versatility makes it particularly valuable for studying proteins that are challenging to crystallize or are inherently dynamic. Unlike mutagenesis, antigen size and complexity do not impact the feasibility or labor intensity of HDX-MS.
- High-resolution: With advanced data analysis methods and high-resolution mass spectrometers, HDX-MS can achieve sub-peptide or even residue-level resolution in some cases (1 to 5 amino acids). This high-resolution capability allows researchers to pinpoint specific regions or residues involved in protein-protein interactions with high precision.
- Structural context and dynamic information: HDX-MS provides valuable structural context by analyzing proteins in three dimensional structures. It can reveal changes in protein conformation and dynamics induced by ligand binding or other environmental factors, providing insights into the mechanisms of protein function and regulation.
Disadvantages and Limitations of HDX-MS Epitope Mapping
Certainly, HDX-MS does have its disadvantages and limitations:
- Specialized equipment and experience: HDX-MS requires specialized equipment, including high-resolution mass spectrometers and automated systems for sample handling and data analysis. Additionally, expertise in mass spectrometry and sophisticated computational tools are needed for data interpretation. This can limit access to the technique for smaller research labs without dedicated resources.
- Optimization challenges: HDX-MS workflows need to be carefully optimized for each protein of interest, particularly for proteins that are heavily glycosylated or intrinsically disordered. Conditions such as pH, temperature, and solvent composition must be precisely controlled to ensure accurate measurement of hydrogen-deuterium exchange rates.
Antibody Epitope Mapping Services
Identifying the epitopes and paratopes of antibodies is crucial in antibody development. Mutagenesis epitope mapping and HDX-MS both benefit from being conducted in parallel to validate epitopes and extract as much information on the candidate antibody and target antigen as possible. However, there are advantages and disadvantages to both epitope mapping techniques, which need to be carefully considered, taking into account the antigen, antibody, stage of development, and available resources.
Rapid Novor offers an HDX-MS service that can accurately identify the binding site of an antibody to its corresponding antigen with high confidence and resolution. Utilizing the state-of-the-art Waters Cyclic IMS-MS and automated sample handling robotics, we achieve high throughput and medium-resolution mapping of epitopes down to 1-5 amino acids.
HDX-MS can also be integrated with AI in silico modeling, significantly increasing throughput and enhancing epitope validation.
Contact our scientists to learn more about epitope mapping using HDX-MS.

Talk to Our Scientists.
We Have Sequenced 10,000+ Antibodies and We Are Eager to Help You.
Through next generation protein sequencing, Rapid Novor enables reliable discovery and development of novel reagents, diagnostics, and therapeutics. Thanks to our Next Generation Protein Sequencing and antibody discovery services, researchers have furthered thousands of projects, patented antibody therapeutics, and developed the first recombinant polyclonal antibody diagnostics.
Talk to Our Scientists.
We Have Sequenced 9000+ Antibodies and We Are Eager to Help You.
Through next generation protein sequencing, Rapid Novor enables timely and reliable discovery and development of novel reagents, diagnostics, and therapeutics. Thanks to our Next Generation Protein Sequencing and antibody discovery services, researchers have furthered thousands of projects, patented antibody therapeutics, and ran the first recombinant polyclonal antibody diagnostics