 Written by María Gerpe, PhD
Written by María Gerpe, PhD
August 13, 2021
Introduction
Amino acid sequencing is essential for understanding protein structure and function. The two most widely used methods for determining amino acid sequences are Edman degradation and mass spectrometry (MS). Each method has distinct features, advantages, and limitations. This article provides a detailed comparison of these techniques, highlighting key pain points associated with each.
Edman Degradation
How Edman Degradation Works
Edman degradation, developed in the 1950s, is a well-established method for sequencing proteins and peptides. The technique focuses on sequentially removing amino acids from the N-terminus of a peptide chain. Each removed amino acid is then identified using chromatographic or electrophoretic methods, such as high-performance liquid chromatography (HPLC). This stepwise process continues until the entire sequence is determined (Figure 1).
Advantages of Edman Degradation
- High accuracy for sequencing small, well-defined peptides.
- Ideal for single, isolated protein samples.
- Provides direct sequence information from the peptide itself.
- Effective for simple, well-characterized proteins in research.
Challenges and Limitations of Edman Degradation
Despite its precision, Edman degradation has several drawbacks:
- Time consuming: Each cycle take approximately 1 hour.
- Limited to shorter peptides: Effective only for peptides under 30 amino acids.
- High sample requirements: Needs around 100 pmol of peptide per analysis.
- Low throughput: Cannot handle multiple samples simultaneously limited to single well-defined peptides.
- N-terminal blockage: Cannot sequencing peptides that have blocked N-terminals with co- or post-translational modifications, such as acetylation. Up to 50% of eukaryotic proteins have N-terminal blockages.
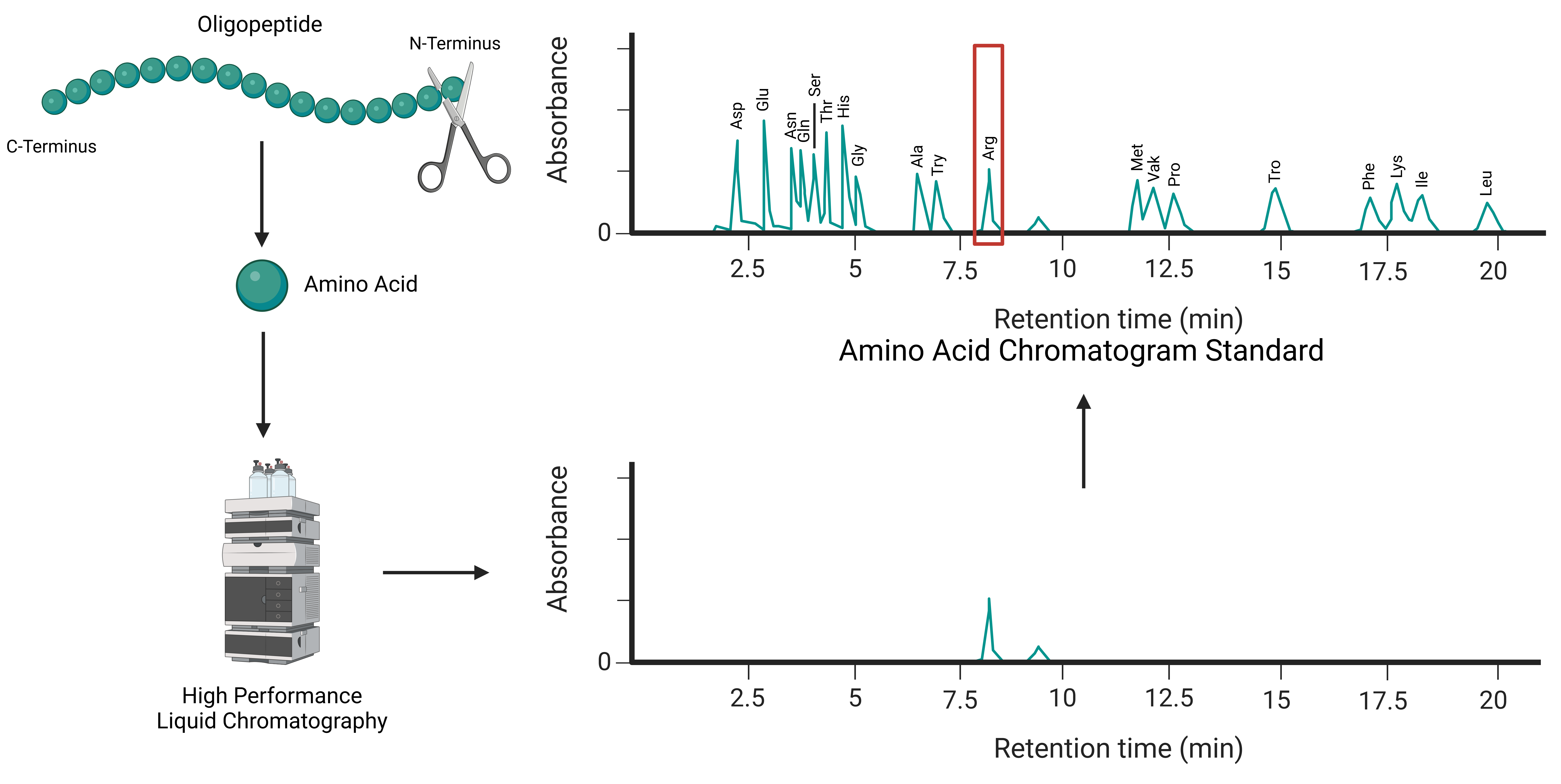
Figure 1: Schematic Diagram of N-terminal Sequence Analysis by Edman Degradation.
Mass Spectrometry
How Mass Spectrometry Works
Mass spectrometry (MS) has emerged as one of the most powerful tools in proteomics. This technique typically involves enzymatic digestion, where a protein is broken down into smaller peptides, usually between 5 to 20 amino acids in length. These peptides are then ionized, and their mass-to-charge ratios (m/z) are measured. By analyzing the mass spectra, researchers can deduce the amino acid sequence of the peptides and ultimately reconstruct the protein sequence.
Strengths of Mass Spectrometry
- High throughput: Can analyze numerous samples simultaneously, make it ideal for large-scale studies.
- Flexibility: Suitable for analyzing a wide range of protein types, from small peptides to large proteins.
- Sensitivity: Recent technological advancements enable detection of even trace amounts of proteins in complex mixtures. MS can be coupled with assays like antibody-based enrichment to offer up to 100,000 times improved sensitivity.
- Post-translational modification (PTM) detection: Can identify protein isoforms and PTMs, providing deeper insights to protein function.
Challenges of Mass Spectrometry
However, mass spectrometry is not without its own set of challenges:
- Dependency on sequence libraries: Successful peptide identification for bottom-up MS often relies on a well-annotated reference database.
- Potential misidentifications: Some genomes have yet to be sequenced, some proteins sequenced via mass spectrometry can be misidentified.
- Detection of isoforms and PTMs: While MS is effective at identifying modifications, it requires additional enrichment strategies (e.g. chromatography) to capture and analyze PTMs fully.
Overcoming Mass Spectrometry Challenges
One solution to the limitations of database-dependent mass spectrometry is de novo sequencing. This approach allows researchers to identify protein sequences without relying on pre-existing databases, bypassing issues related to incomplete or absent reference libraries. Liquid chromatography-tandem mass spectrometry (LC-MS/MS), combined with machine learning algorithms, enables de novo sequencing, where data from the mass spectrometer is directly used to infer the protein sequence (Figure 2).
- Advantages of de novo sequencing:
- No need for reference database
- Can analyze previously uncharacterized proteins
- Uses machine learning algorithms to enhance sequence accuracy.
- Helps detect novel modifications and proteins isoforms and variants.
Despite its potential, de novo sequencing is still not widely available due to its complexity and the need for advanced machine learning integration in lab processes.
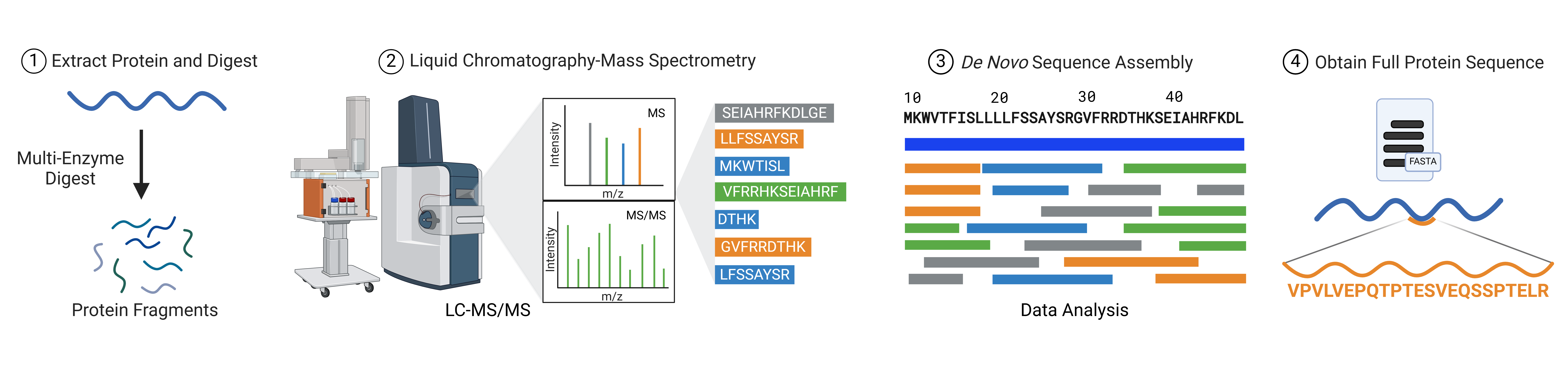
Figure 2: Workflow of de novo protein sequencing using tandem mass spectrometry combined with bioinformatics algorithms.
Summary
Finding the correct technique for amino acid sequencing can be challenging. Edman degradation and mass spectrometry are two popular methods with each one possessing specific pain points. De novo protein sequencing by tandem mass spectrometry is a great tool to bypass these pain points. The latter type of amino acid sequencing technology continues to facilitate antibody discovery and development.
Want to learn more about our antibody sequencing services and antibody discovery services? Contact us for more information.
References
1 Alfaro, J. A. et al. The emerging landscape of single-molecule protein sequencing technologies. Nat Methods 18, 604-617, doi:10.1038/s41592-021-01143-1 (2021).
2 Hughes, C., Ma, B. & Lajoie, G. A. De novo sequencing methods in proteomics. Methods Mol Biol 604, 105-121, doi:10.1007/978-1-60761-444-9_8 (2010).

Talk to Our Scientists.
We Have Sequenced 10,000+ Antibodies and We Are Eager to Help You.
Through next generation protein sequencing, Rapid Novor enables reliable discovery and development of novel reagents, diagnostics, and therapeutics. Thanks to our Next Generation Protein Sequencing and antibody discovery services, researchers have furthered thousands of projects, patented antibody therapeutics, and developed the first recombinant polyclonal antibody diagnostics.
Talk to Our Scientists.
We Have Sequenced 9000+ Antibodies and We Are Eager to Help You.
Through next generation protein sequencing, Rapid Novor enables timely and reliable discovery and development of novel reagents, diagnostics, and therapeutics. Thanks to our Next Generation Protein Sequencing and antibody discovery services, researchers have furthered thousands of projects, patented antibody therapeutics, and ran the first recombinant polyclonal antibody diagnostics