March 23, 2021
Jessica Barber, MPH*,1, Yuning Wang, PhD#,1
*Author
#Reviewer
1Rapid Novor, Inc.
Abstract
In vitro diagnostics (IVDs) are one of the most commonly used tools to diagnose conditions and guide treatment decisions, and are often considered the “silent champion” of healthcare. They work by detecting the absence or presence of particular markers or by measuring the concentration of analytes or specific substances. Some examples include measuring genetic mutations or immune responses to an infection. Immunodiagnostics are one of the most common types of IVD; they exploit antibody-antigen reactions to diagnose a variety of diseases, including infections, cardiac diseases, neurodegenerative disorders and cancers. Antibodies directly affect the reliability and repeatability of immunodiagnostics. Therefore, it is critical to rigorously assess and validate antibodies used for IVD purposes prior to clinical use as clinicians often rely on them to make treatment decisions. Across disciplines, antibody validation relies on molecular biology techniques that exclude verification of the sequence of the final protein sequence. This review highlights the challenges associated with diagnostic test development, and identifies potential solutions, including the use of protein sequencing.
Key Takeaways
- For patent eligibility or infringement issues, access to the antibody sequence is becoming a critical piece of information
- Batch-to-batch variability of IVD antibodies resulted in $500,000 wasted for two researchers
- Verification of the protein sequence of antibodies can be a vital step in ensuring antibody validation
- Protein sequencing can also be used to enhance discovery and development of IVD antibodies
Introduction
In vitro diagnostics (IVDs) are one of the most commonly used tools to diagnose conditions and guide treatment decisions, and are often considered the “silent champion” of healthcare1. They work by detecting the absence or presence of particular markers or by measuring the concentration of analytes or specific substances. Some examples include measuring genetic mutations or an immune response to an infection2.
Although IVDs currently influence two-thirds of clinical decisions, they only account for 2% of healthcare spending1. It has been estimated that the IVDs market will reach $96.0 billion by 20253, with previous reports estimating US$73.9 billion by 2020, up from US$61.9 billion in 20164.
The World Health Organization (WHO) identifies IVDs as being particularly useful as the primary care setting where laboratories are not available5, due to the availability of conducting the tests in various locations such as at patient’s homes. IVDs are regularly used by clinicians to monitor a person’s overall health, including relapse detection, treatment efficacy, and disease prevention.
Immuno-diagnostics is one of the most common IVD streams that adopts antibody-antigen reactions for diagnostic purposes. These tests can be used to diagnose a variety of diseases, including infections, cardiac diseases, neurodegenerative disorders and cancers. Antibodies (Abs) are one of the most frequently used analyte specific reagents for IVDs and directly affect the reliability and repeatability of the tests. Therefore, it is critical to rigorously assess the Abs’ affinity and specificity to the target molecules prior to use.
As patients and clinicians may depend on the diagnostic results to make treatment decisions, it is critical these tests are reliable2. In this article, our team wishes to bring to light some of the challenges associated with diagnostic test development, and identify potential solutions.
The State of IVDs
A Push for Rigorous Regulation & Validation
In the United States, IVDs are regulated by the Food and Drug Administration (FDA) as medical devices under the Medical Device Amendments of 1976. This bill requires that manufacturers must submit studies confirming the test’s accuracy and usefulness in diagnosing a particular condition before bringing it to market1. In order to be cleared by the FDA, IVDs must demonstrate safety and effectiveness through both analytical and clinical validation. For analytics, the focus is on ensuring the test is correctly and reliably measuring a specified analyte. Whereas, for clinical validation, the focus is on determining whether it can accurately identify a particular clinical condition in a given patient2.
In European markets, including EU member states and EFTA states, IVDs are regulated by the In Vitro Diagnostic Regulation (IVDR) as of May 25, 2017. Beginning May 26, 2022 medical device manufacturers will be required to comply with the new IVDR in order to be placed in the European market6. With this new regulation, manufacturers will need to provide more rigorous clinical evidence including conducting more clinical performance studies and providing more evidence of safety and performance according to the device’s assigned risk class. Additionally, the IVDR will require more stringent documentation while implementing unique device identification for better traceability and recall6.
Biological Sequences: A Tool for Traceability in Patent & Commercial Applications for IVD
Although biological inventions do not require for the DNA or protein sequences to be disclosed, within the context of patent eligibility or infringement issues, their structure and function value is gaining more importance7. Further, the biological sequence information can provide an additional level of traceability and reproducibility.
Biological sequences are being viewed both legally and practically within patent applications, and as such, there are specific guidelines to follow. Notably, due to their function, biological sequences can only be determined with computer-aided technology; highlighting the importance of the sequences’ submitted format7. The most common format used is FASTA, which is a text-based format8.
Further, at the manufacturing level, there has been an industry push to hold vendors accountable to defining their antibody reagents based on unique identification numbers, known as Research Resource Identifiers (RRIDs). This will lead to a considerable increase in research traceability and reproducibility as scientists will be provided access to a database with information about what reagents to use and how they perform across systems, including original catalog numbers and vendor names.
Only 11% of landmark preclinical studies can be replicated
Although most of the reagents used in clinical trials cleared by the FDA or European Medicines Agency are considered reliable, due to the lack of industry regulation including quality control protocols, indefinite reproducibility remains to be a concern. This lack of vigilance has resulted in the widespread unintended use of cross-reactive antibodies, inaccurate data sets, and catastrophic waste of funds and time; ultimately significantly slowing progress in medical science9.
It is estimated that by 2019 global spending on antibodies rose to US$3.4 billion9, yet, one study suggests US$1.7 billion could have been wasted. Within this 2008 study, 6,000 commercial antibodies were tested and only half could successfully recognize their specified targets10. Further, within C. Glenn Begley’s study of 53 landmark preclinical studies, only 6 results were able to be replicated.
What is Damaging and Invalidating Clinical IVD Trials?
The most frequent challenge for Ab-based research is when the Abs have not been experimentally verified with rigorous scrutiny prior to use. This is often due to the preconceived notion that if researchers purchase from reputable vendors or rely on industry citations, they presume validity and specificity has been pre-established. As a result, these poorly-characterized Abs can suffer from cross-reactivity, meaning they exhibit high affinity toward other antigens, causing serious trouble in users’ research.
The cross-reactivity can happen in both polyclonal and monoclonal antibodies (mAbs). Polyclonal antibodies (pAbs) often show a higher degree of cross-reactivity due to their heterogeneous nature that enables them to recognize multiple epitopes. mAbs, although considered highly specific, can also exhibit off-target binding if not well validated. In a study conducted by Rapid Novor in 2019, among 80 research-purpose mAbs analyzed, a significant portion (14%) were shown to have a second light chain present11. This is likely resulted from genome instability of hybridoma cell lines producing the mAbs12.
The damage incurred by the use of improperly validated Abs becomes worse when such reagents find their way into the clinic as established tools for biomarker detection, thus damaging and invalidating costly clinical trials9.
Take for example the following Abs that were used to identify therapeutically relevant clinical biomarkers. Despite all of these Abs being regularly cited in industry publications and journals for their respective use cases, all were shown to exhibit cross-reactivity resulting in significant sums of research resources being lost.
Table 1 Lists some cross-reactive antibodies that were incorrectly used to identify therapeutically relevant clinical biomarkers. Originally displayed at: https://escholarship.org/uc/item/98r4z3x7
| Target | Antibody IDs | Biomarker | Cross-reactions |
| EpoR (EPOR) | M20 and C20 | Tumor cells | HSP70 |
| ER-β (ESR2) | 12 out of 13 | Breast cancer | WDCP, POU2F1, multiple |
| HER2 (ERBB2) | 2 out of 3 | Breast cancer | HER4 |
| ERCC1 | 8F1 | Prognostic | CCT-alpha |
| CDK1 | A17 | Cancer | Cep152 |
Thus, using Abs as tools for detection of biomarkers can be potentially fatal if IVD manufacturers have not fully verified their Abs with rigorous scrutiny. The unfortunate reality is, it’s no longer sufficient to rely solely on vendor’s quality assurance protocols or scientific publications, it is necessary to independently assess and verify candidates.
How Protein Sequencing Safeguards Assays & Avoids Redevelopment
Ab-based diagnostic platforms can represent a large sum of a company’s annual revenue, therefore validation is a necessary process – between identifying and sourcing different mAbs, testing them experimentally, and choosing the final one to use long term. Through sequencing validated antibodies, it is possible to ensure that validation expenses are not incurred multiple times.
Take these Canadian researchers as a prime example. Dr. Prassas and Dr. Diamandis spent $500,000 and 2 years of research hours only to uncover their commercial assay for CUZD1 was actually measuring CA12513. These researchers predetermined that manufacturer quality assurance was sufficient and additional experimental verification was unnecessary. Unfortunately, the reality is that investigators around the world should be aware that certain suppliers are releasing with fast pace ELISA kits of questionable quality13.
Researchers at NeuroDex opted to ensure that once they identified their candidates, that their research would remain reproducible. They did this through obtaining the protein sequence. Their team is developing an exosome-based noninvasive diagnostic tool for Alzheimer’s disease and other neurological disorders. The platform is based on neuron-derived exosome isolation technology whereby neuron-specific Abs are used to pull down and recover the neuronal exosome particles from plasma. As this platform is dependent on the quality and reproducibility of the Ab that recognizes and binds particular exosome surface markers, ensuring they have a reliable supply with consistent performance is of paramount importance. Obtaining the de novo protein sequence has enabled NeuroDex to recombinantly synthesize their valuable antibody, ensuring continuous and reliable supply.
Optimizing Target Discovery & Development of IVD
Target discovery is the identification and early validation of disease-modifying targets. However, this discovery should extend well beyond the basic research of the disease relevance. Notably, target discovery should encapsulate all structural information including all protein interactions. As the success and approval of IVDs is directly related to their ability to reliably and accurately measure specific analytes, fully characterizing the protein of interest is a crucial first step.
When biomarkers have been improperly identified or lack sufficient experimental verification, tests can have unreliable results leading to patient harm. Take both LabCorp and Theranos, who independently marketed diagnostic tests that had inaccurate results which harmed the patients.
In 2008, LabCorp’s OvaSure test was marketed as a test capable of detecting ovarian cancer. Although the test initially showed promising results, it was identified that uncertainty pertaining to the test’s validity was downplayed. This resulted in patients undergoing irreversible and life-altering surgery based on faulty test results. Then again in 2012, another diagnostic company, Theranos, unveiled a new microfluidics test that they claimed was capable of identifying a range of conditions with only a few drops of blood. Yet, ultimately the CMS invalidated all of their test results due to false claims. This was however after multiple patients underwent unnecessary treatments as a result of false tests; resulting in a class-action lawsuit2.
These are just a few of the documented stories where the consequences of improperly validated antibodies are known. In an effort to move towards better outcomes for both patients, researchers, and stakeholders, it is imperative that biomarkers and reagents be extensively validated before use.
Antibody protein sequencing is an important part of validation and is growing in popularity for the following applications:
- Researchers can confirm the sequence of synthesized or procured antibodies to validate identity, prior to expensive and time consuming downstream work.
- If an antibody is not binding as expected, knowledge of the antibody protein sequence provides essential information in troubleshooting binding interactions.
- In early stage development, if you are trying to find or produce several distinct antibodies for a particular antigen, it is useful to compare sequences of several binders to ensure they are not too similar.
How to Secure Indefinite Reproducibility of IVDs
Antibody protein sequencing provides researchers with a solution to rigorously test their materials prior to investing countless hours and resources into unverified products. De novo sequencing works by determining the mass difference between two fragment ions from tandem mass spectra. By first employing an enzyme digest the reagent is fragmented into smaller peptides, which can then be analysed by LC-MS/MS. From here, the spectral data can be analyzed manually, or preferably, by an automated software in order to assemble the full amino acid sequence (Figure 1).
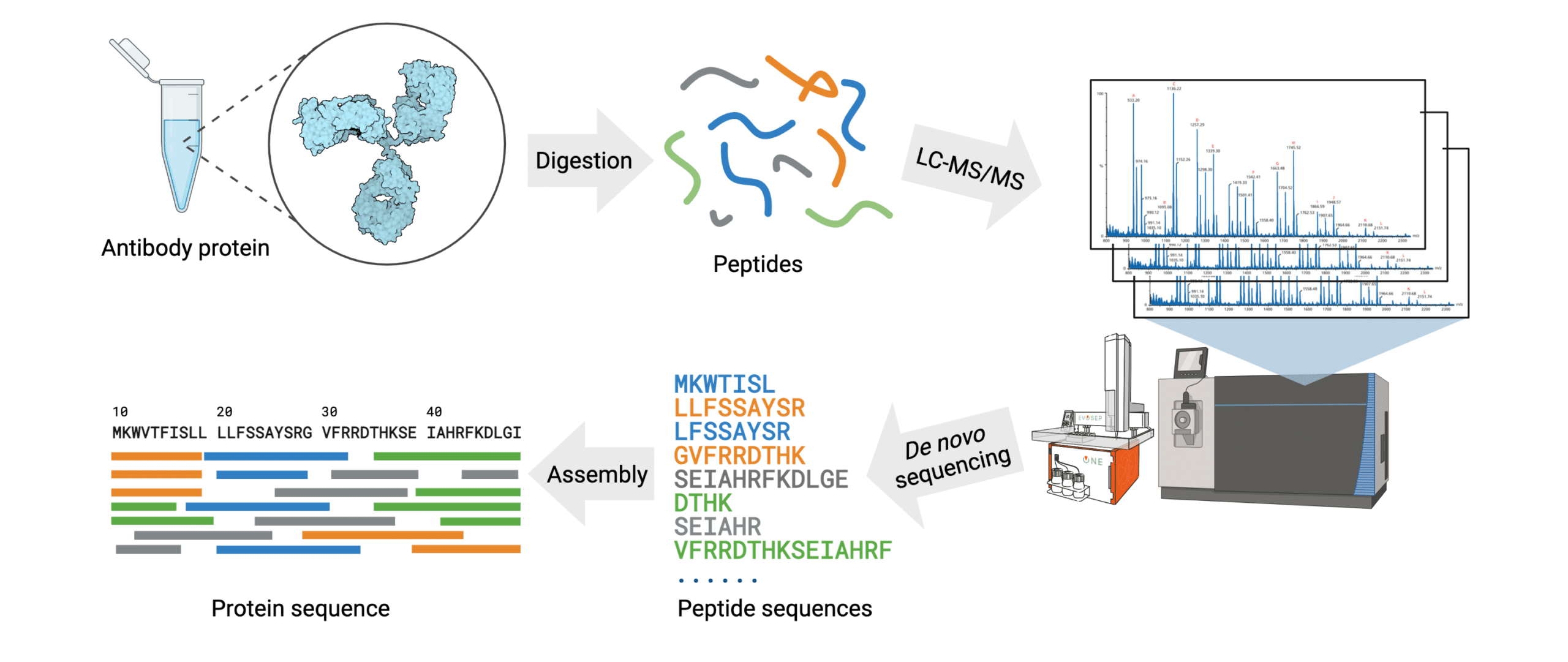
Figure 1. Workflow of the REmAb® de novo sequencing platform.
Knowing the full amino acid sequence allows researchers to recombinantly express the Ab, enabling indefinite reproducibility with predictable biologic activity. Furthermore, antibody protein mass and sequence confirmation are valuable in antibody production quality control.
The REmAb® Advantage
Rapid Novor’s proprietary de novo sequencing platform, REmAb®, successfully determines the primary amino acid sequences of Abs without requiring the original hybridoma cell lines or prior knowledge of DNA sequences. By directly analyzing the Ab protein, this technology provides a solution to resurrect lost hybridomas, re-engineer proteins, and re-establish important Abs with high consistency.
By employing their proprietary Big Data-driven machine learning algorithm, Rapid Novor assembles and generates the full Ab protein sequence within 1-2 weeks requiring only 100 µg of starting material. This platform guarantees an average of at least 30X coverage for each amino acid, resulting in 100% accuracy.
Rapid Novor’s team has successfully de novo sequenced more proteins than any other organization in the world. This expertise and experience has allowed them to further push the boundaries of proteomics, leading to advancements in the likes of oligoclonal and polyclonal sequencing.
REmAb® Strengthens IVD development
- When developing the antibody, REmAb® can make sure it is pure and does not contain any additional chains/minor variants. This can help avoid potential cross-reactivity.
- If an antibody has been developed with the desired specificity and binding affinity, REmAb® can determine the sequence of the Ab to potentially patent it as a part of the assay and thus protect intellectual property.
- Aids with the quality control of antibody candidates.
- Accurately troubleshoots when development is experiencing inconsistency.
- Enables proof of concept for kit development.
- Avoids assay redevelopment costs.
- Introduces the ability to bring development in-house.
- Ensures an indefinite supply of Abs with predictable biologic activity, thus eliminating lot-to-lot variability.
References
1. Making future breakthroughs possible. Risk-based rules for in vitro diagnostics. 2021. [cited 2021 Mar 9]; Available from: https://www.roche.com/about/business/diagnostics/value-of-ivds.htm
2. What Are In Vitro Diagnostic Tests, and How Are They Regulated? [Internet]. [cited 2021 Mar 4]. Available from: https://pew.org/2JbtGcr
3. ReportLinker. The global in-vitro diagnostics market size is projected to reach USD 96.0 billion by 2025 from USD 84.5 billion in 2020, at a CAGR of 2.6% [Internet].
4. GlobeNewswire NewsRoom. 2020 [cited 2021 Mar 4]. Available from: http://www.globenewswire.com/news-release/2020/11/26/2134368/0/en/The-global-in-vitro-diagnostics-market-size-is-projected-to-reach-USD-96-0-billion-by-2025-from-USD-84-5-billion-in-2020-at-a-CAGR-of-2-6.html
5. A close look at in vitro diagnosis. [cited 2021 Mar 4]; Available from: https://www.nature.com/articles/d42473-018-00236-4
6. WHO | Laboratory and in vitro diagnostics [Internet]. WHO. World Health Organization; [cited 2021 Mar 4]. Available from: http://www.who.int/in-vitro-diagnostic/en/
7. EU In Vitro Diagnostic Medical Device Regulation [Internet]. www.tuvsud.com. [cited 2021 Mar 4]. Available from: https://www.tuvsud.com/en/industries/healthcare-and-medical-devices/medical-devices-and-ivd/medical-device-market-approval-and-certification/eu-in-vitro-diagnostic-medical-device-regulation
8. Jefferson OA, Köllhofer D, Ajjikuttira P, Jefferson RA. Public disclosure of biological sequences in global patent practice. World Patent Information. 2015 Dec 1;43:12–24.
9. New tools and advancements in biological sequence searching | Lexology [Internet]. [cited 2021 Mar 4]. Available from: https://www.lexology.com/library/detail.aspx?g=48d5c804-0af6-4de1-a375-5e09c1ad3e06
10. Voskuil JLA, Bandrowski A, Begley CG, Bradbury ARM, Chalmers AD, Gomes AV, et al. The Antibody Society’s antibody validation webinar series. mAbs. 2020 Jan 12;12(1):1794421–179441794421.
11. Bradbury A, Plückthun A. Reproducibility: Standardize antibodies used in research. Nature News. 2015 Feb 5;518(7537):27.
12. McDonald, Z. et al. Studying the Prevalence of Secondary Light Chains in Research Purpose Monoclonal Antibodies with MS-Based De Novo Protein Sequencing. ASMS 2018 San Diego, MP 063. Available from: https://www.rapidnovor.com/research/prevalence-of-secondary-light-chains/
13. Bradbury, Andrew R M et al. “When monoclonal antibodies are not monospecific: Hybridomas frequently express additional functional variable regions.” mAbs vol. 10,4 (2018): 539-546. doi:10.1080/19420862.2018.1445456
14. Prassas, I., Diamandis, P. Translational researchers beware! Unreliable commercial immunoassays (ELISAs) can jeopardize your research. Editorial. Clin Chem Lab Med (2014);52(6):765-766. DOI: 10.1515/cclm-2013-1078

Talk to Our Scientists.
We Have Sequenced 10,000+ Antibodies and We Are Eager to Help You.
Through next generation protein sequencing, Rapid Novor enables reliable discovery and development of novel reagents, diagnostics, and therapeutics. Thanks to our Next Generation Protein Sequencing and antibody discovery services, researchers have furthered thousands of projects, patented antibody therapeutics, and developed the first recombinant polyclonal antibody diagnostics.
Talk to Our Scientists.
We Have Sequenced 9000+ Antibodies and We Are Eager to Help You.
Through next generation protein sequencing, Rapid Novor enables timely and reliable discovery and development of novel reagents, diagnostics, and therapeutics. Thanks to our Next Generation Protein Sequencing and antibody discovery services, researchers have furthered thousands of projects, patented antibody therapeutics, and ran the first recombinant polyclonal antibody diagnostics