Thierry Le Bihan1, Paul Taylor1, Zac McDonald1, Qixin Liu1, Jianqiao Shen1, Kathleen Gorospe1, Xin Xu1, Chris Hosfield2, Bin Ma1,3
1Rapid Novor, Inc, Kitchener, Ontario, Canada
2Promega Corporation, Madison, WI
3University of Waterloo, Waterloo, Ontario, Canada
Abstract
In this study, we conducted a large-scale statistical analysis of protein sequencing data from samples digested with multiple proteases to understand the impact of using different combinations of proteases to improve the depth of sequence coverage in the application of de novo protein sequencing. MS data for 166 monoclonal antibodies were compiled for use in this study. Each antibody protein sample was digested separately with different proteases and analyzed by LC-MS/MS. The new Pro/Ala (A/P) protease was tested to characterize NIST and MAB04HC antibody standards. De novo peptide sequencing was performed with Novor search algorithm. Protein sequences were assembled using REmAb®. The assembled mAbs demonstrate that a combination of existing proteases with orthogonal activities significantly increases confidence scores in de novo protein sequencing; however, there is a need for new proteases targeting specific amino acid(s) (a.a.) or a.a. sequences to increase antibody sequencing accuracy.
Key Takeaways
- A combination of proteases can maximize coverage during LC-MS/MS-based sequencing
- Other orthogonal approaches also contribute to an increase in accuracy for de novo protein sequencing
- REmAb®’s de novo protein sequencing established protocol includes orthogonal and protease cocktails optimized to deliver highly accurate and full-coverage protein sequences
Introduction
Sample preparation for complete LC-MS/MS sequencing of a pure protein sample differs substantially from the preparation used for protein identification in complex samples. To sequence a protein de novo, the experimental design should aim to identify each amino acid (a.a.) multiple times within different peptides, instead of relying on only a few peptides per protein for complex protein mixtures. Efforts to increase protein sequence coverage typically rely on using multiple proteases to digest the protein. In this study, we conducted a large-scale statistical analysis of protein sequencing data from samples digested with multiple proteases to understand the impact of using different combinations of proteases to improve the depth of sequence coverage in the application of de novo protein sequencing. The data presented here can help guide the choice of proteases for maximum coverage during protein sequencing.
Materials & Methods
Proteases and Target Proteins:
Trypsin, Lys-C, Chymotrypsin, Pepsin, A/P, and recombinant Asp-N proteases Promega (Promega, WI, US) were used to digest NISTmAb humanized IgG1 monoclonal antibody RM8671 (National Institute of Standards and technology, U.S. department of Commerce), monoclonal mAb04 and yeast extracts (Promega, WI, US).
LC-MS/MS:
Protein lysates were analyzed with an Orbitrap Fusion™ Series Tribid™ instrument (ThermoFisher Scientific, CA, US) coupled to the LC Evosep One (Evosep, Denmark) in both HCD and ETD mode. ETD spectra were acquired with three different collisions energies.
Data analysis:
De novo peptide sequencing was performed with Novor search algorithm and the protein sequences were assembled with REmAb® (Figure 1). Yeast complex samples were analyzed with Search GUI-3.3.13 and PeptideShaker-1.16.37 using no enzyme restriction, 25ppm with protein, peptide and 1% psm FDR, while considering a combination of X! tandem, MS-GF+ and Comet. Sequence motif analysis was performed using SeqtoLogo.
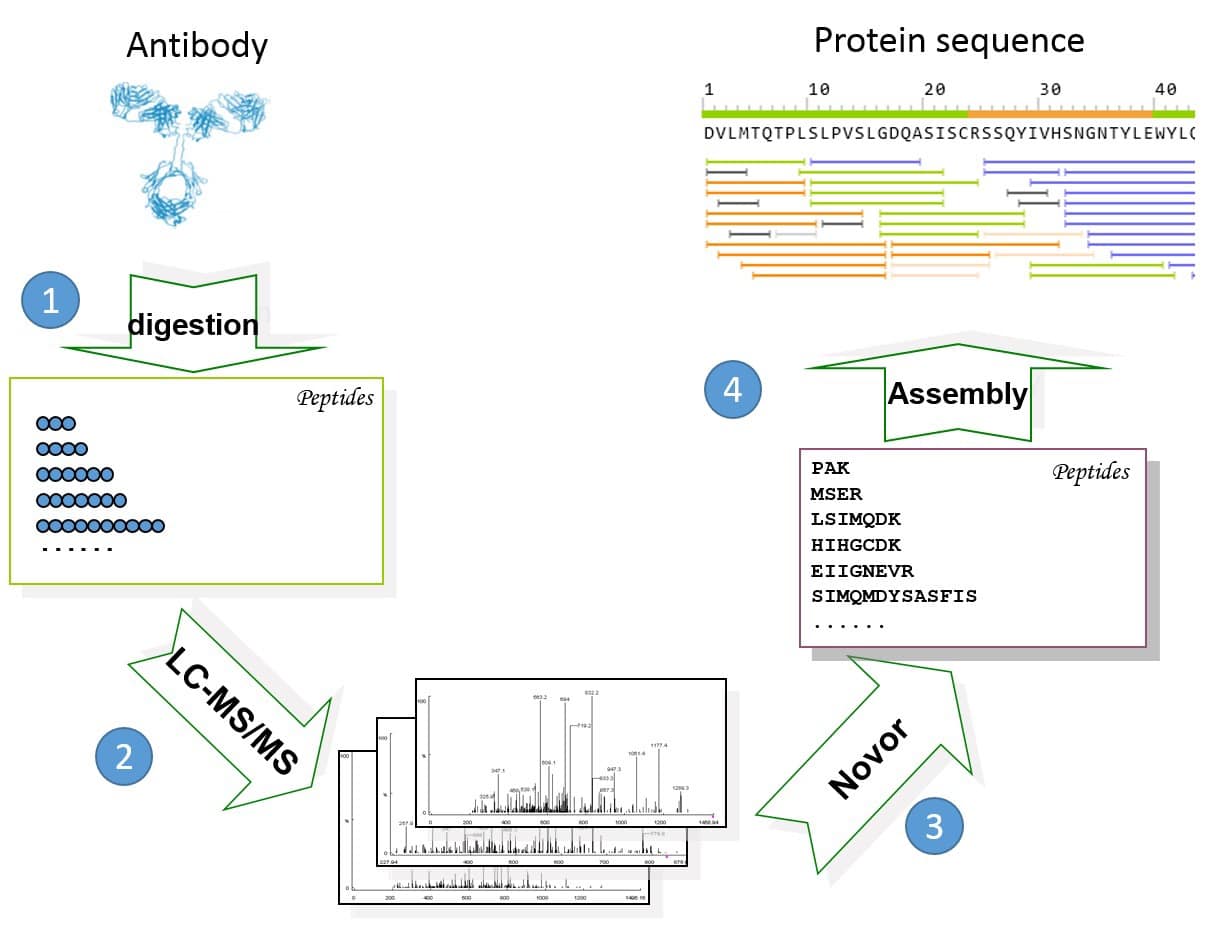
Figure 1. General workflow of REmAb® includes (1) multiple enzyme digests, (2) mass spec, (3) de novo peptide sequencing, and (4) sequence assembly.
Results
The A/P Protease can be used to further validate protein sequencing
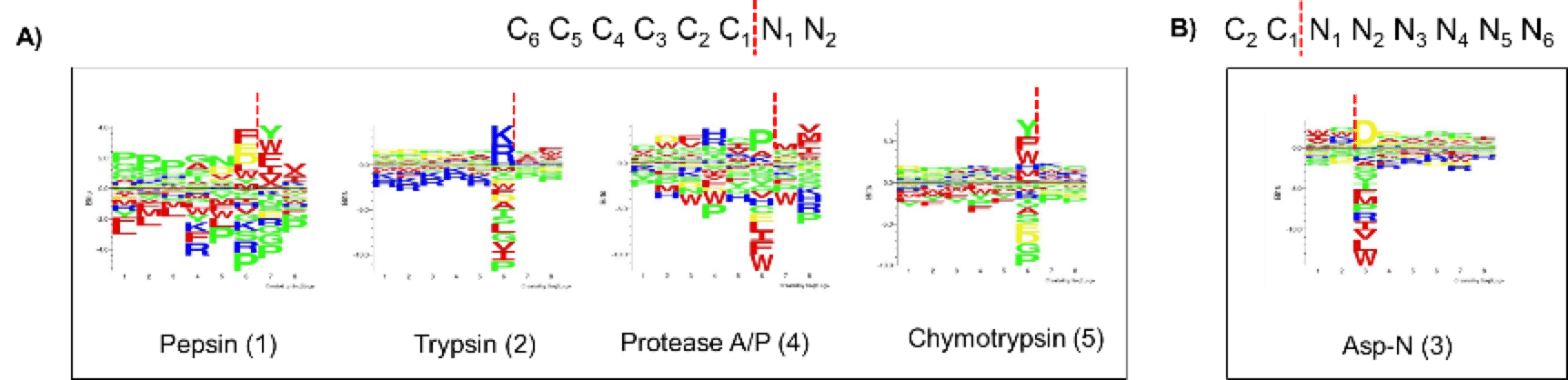
Figure 2. Seq2Logo sequence motif logo displaying analysis of C-ter (A) and N-ter (B) fragments of yeast protein extract digested with five different proteases. The numbers in brackets refer to the LC-MS/MS run order. The red vertical dotted lines indicate cleavage sites. Four of these proteases are C-ter amino acid-specific: Trypsin, pepsin, chymotrypsin and Ala/Pro (A/P), and one is N-ter-specific: Asp-N. Our data highlight that pepsin preferentially cleaves the C1 N1 site while chymotrypsin mainly targets the C1 site. Trypsin, pepsin, chymotrypsin and Asp-N proteases show proline-related inhibition in the vicinity of the cutting site; in contrast, the Ala/Pro protease has a preference for the C1 Proline. These findings demonstrate that the protease A/P can be used to efficiently complement protein sequencing data.
Use of diverse proteases facilitates maximum coverage of CDR3
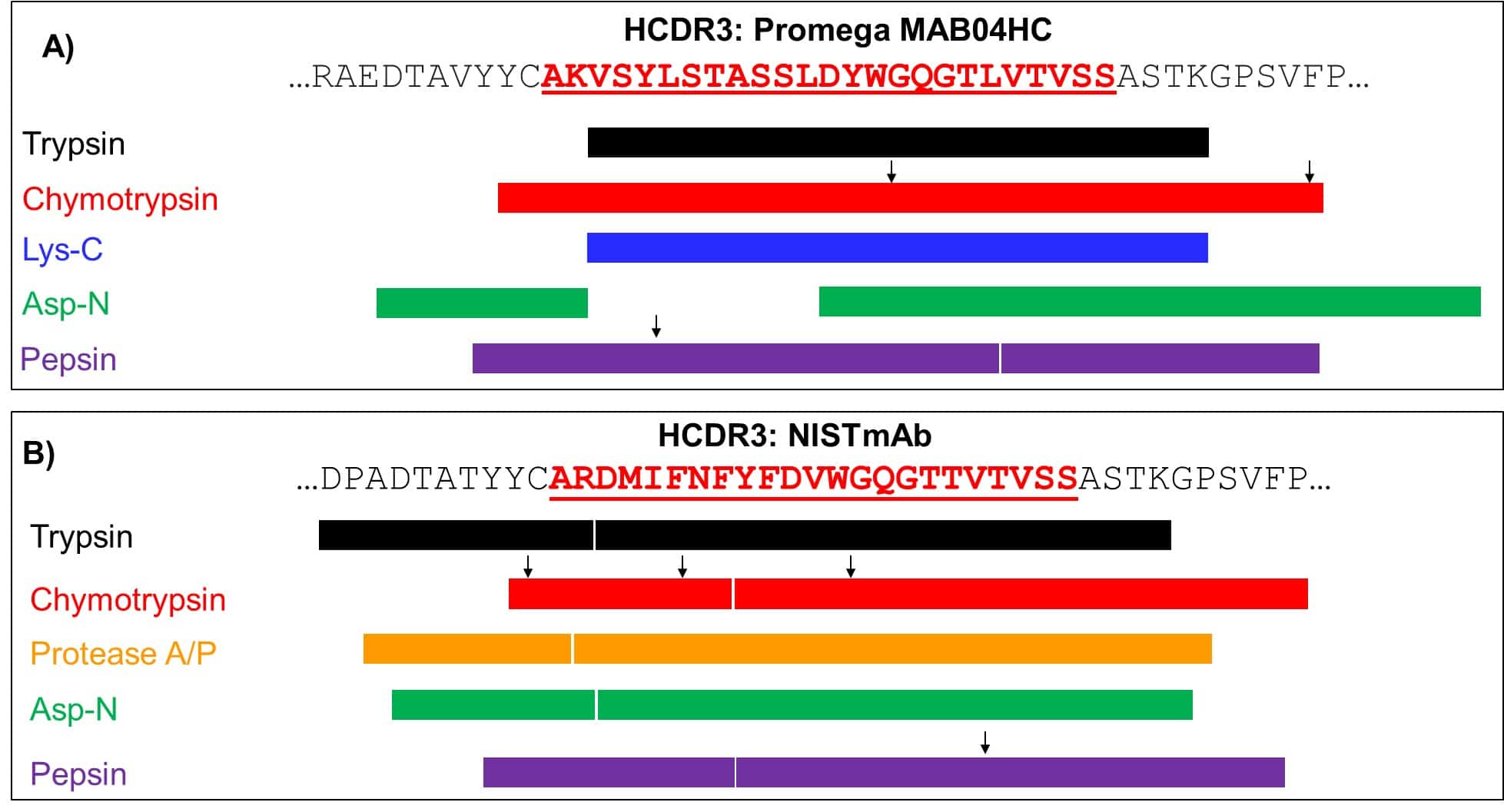
Figure 3. Graphical representation showcasing the overall sequence coverage spanning the HCDR3 peptide sequence for two different standard monoclonal antibodies, Mab04HC (A) and NIST (B). Depicted by different colors, different proteases (Trypsin, Chymotrypsin, A/P, Asp-N and Pepsin) were utilized to cleave the antibodies to generate fragments of different sizes. The black arrows highlight the different cleavage sites found within a given sequence. A nearly complete coverage of both HCDR3s (bolded and underlined in red) is obtained after cleavage by each protease. The advantage of using different proteases is the resulting sequence overlap that can be used to facilitate de novo protein assembly.
Protease combinations are important to achieve maximum coverage
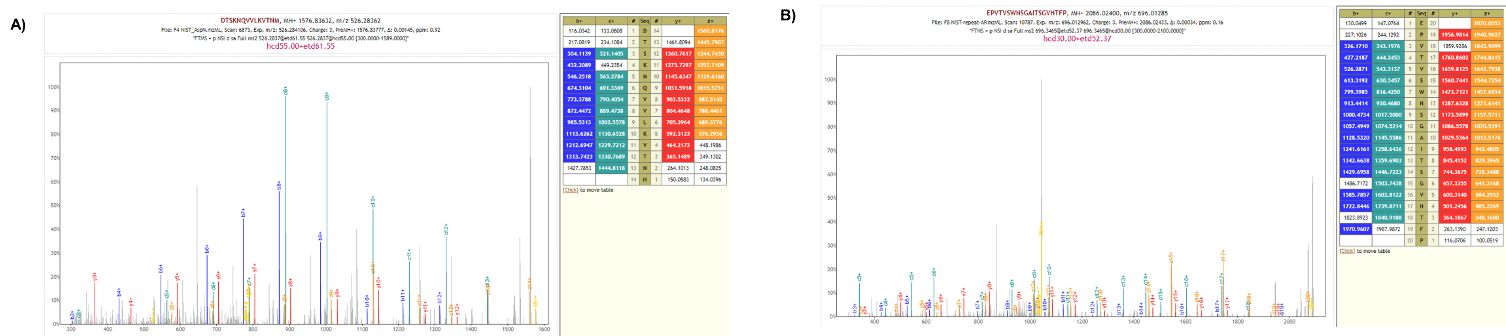
Figure 4. MS/MS spectra and annotation using de novo software Novor from sample data resulting from Asp-N protease digestion (A) and protease P/A (B) of the NIST antibody. De novo sequencing performed using Novor based on b, c, y and z ions (from HCD, ETD spectra). Despite their non tryptic nature, a good coverage of both N- and C-terminal ends is observed. The conserved region of the NIST antibody was estimated to be covered at 89% using trypsin, 70% using chymotrypsin, 91% using the protease A/P, 73% using Asp-N and 70% using pepsin. We found those values vary from one antibody to another one.
The use of many different proteases translates to wider coverage
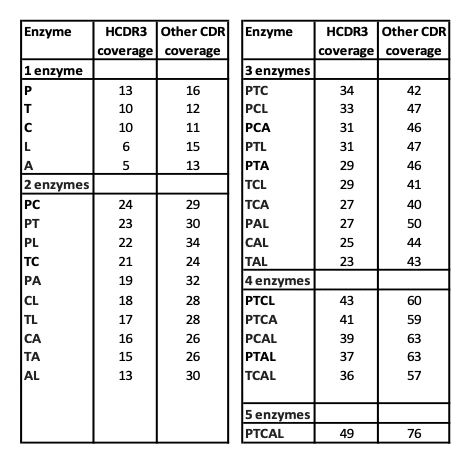
Table 1. CDR coverage analysis of 166 antibodies.
The median depth of coverage for the HCDR3 region and the remaining portion of the variable regions are listed in the following table for each combination of the proteases used in this study. The first column lists the protease combination, where each protease is represented by a single letter code: P (Pepsin), T (Trypsin), C (Chymotrypsin), A (Asp-N), and L (Lys-C). The second and third columns list the median depths of coverage of the HCDR3 and the other variable portions, respectively. The “depth of coverage” for an amino acid is defined as the number of unique PSMs covering the amino acid. Repeated MS/MS scans of the same precursor were counted as a single PSM. We compared the media depth of coverage achieved by different combinations of proteases by examining all amino acids from 166 antibodies. As expected, a greater number of different proteases increase coverage. In all cases, the HCDR3 is less covered than any other variable region. Surprisingly, when a limited number of proteases is employed, pepsin seems to significantly contribute to improve amino acid coverage. We propose that this is most likely associated with the generation of a greater number of peptides with miss-cleaved sites and therefore a wider pool of different peptides.
Conclusions
Successful de novo antibody sequencing depends on full coverage of the protein of interest that is best achieved through repeated identification of amino acid in different peptides with overlapping sequences; different proteases with different cleavage site rules can be used to make de novo antibody protein sequencing a success.
Less specific proteases such as chymotrypsin and pepsin generate more overlapping peptides than more specific proteases such as trypsin and Lys-C. This explains why studies have shown that employing a lower number of proteases can result in a higher amino acid coverage.
However, we observed that the presence of proline can result in inefficient cutting by trypsin, chymotrypsin, Asp-N, and pepsin. The recently commercially available A/P protease is capable of cutting peptides at C1 proline sites. Our findings show that the A/P protease can be used as a complementary tool to de novo protein sequencing. Particularly, in the case of antibody sequencing, additional proteases will be important for targeting conserved amino acid or specific motifs to facilitate their sequencing. We found that this was especially important for the CDR3 region, which is often a difficult-to-sequence antibody area.
This case study was adapted, with permission, from Le Bihan, T., Taylor, P., McDonald, Z., Liu, Q., Shen, J., Gorospe, K., Xu, X., Hosfield, C., Ma, B. (2019). Increased De Novo Protein Sequencing Coverage with Optimal Protease Cocktail. ASMS 2019 Atlanta, TP 020, with permission.

Talk to Our Scientists.
We Have Sequenced 10,000+ Antibodies and We Are Eager to Help You.
Through next generation protein sequencing, Rapid Novor enables reliable discovery and development of novel reagents, diagnostics, and therapeutics. Thanks to our Next Generation Protein Sequencing and antibody discovery services, researchers have furthered thousands of projects, patented antibody therapeutics, and developed the first recombinant polyclonal antibody diagnostics.
Talk to Our Scientists.
We Have Sequenced 9000+ Antibodies and We Are Eager to Help You.
Through next generation protein sequencing, Rapid Novor enables timely and reliable discovery and development of novel reagents, diagnostics, and therapeutics. Thanks to our Next Generation Protein Sequencing and antibody discovery services, researchers have furthered thousands of projects, patented antibody therapeutics, and ran the first recombinant polyclonal antibody diagnostics