June 30, 2021
 Yuning Wang, PhD*,1, Maria Gerpe, PhD#,1
Yuning Wang, PhD*,1, Maria Gerpe, PhD#,1
*Author
#Reviewer
1Rapid Novor, Inc.
Abstract
Anti-drug antibody (ADA) assays are critical to assess the clinical efficacy and safety of a biological drug and rely on control reagents that mimic the ADA response to the biological drug being tested. These positive controls typically consist of animal-derived pooled polyclonal antibodies or human monoclonal antibody reference panels against the target protein drug. However, the existing methods to develop positive controls are prone to many challenges such as low accuracy, poor reproducibility, finite amounts, and lengthy production. In this review, we provide an overview of the current state of ADA assays and discuss how next generation protein sequencing can mine anti-drug antibodies from the vast polyclonal response to said target drug. Finally, we highlight how a polyclonal antibody sequencing platform that incorporates recombination production can optimize and strengthen ADA assay development by overcoming the above challenges.
Key Takeaways
- All ADA approaches involve the use of positive control antibodies to determine assay sensitivity, adverse effects, and drug neutralization or tolerance
- Global regulatory agencies recommend the use of antibody reference panels
- Obtaining the sequence of antibodies from the polyclonal ADA response would ensure reproducible standards that would last indefinitely
- Current, production methods are dependent on cells and/or nucleic material
- Polyclonal antibody sequencing technology is a novel tool that facilitates the rapid discovery of antibody standards for ADA assays by tapping directly into the circulating proteins for a more faithful recapitulation of the relevant ADA response
Anti-drug antibodies: a brief primer
What is an anti-drug antibody?
While therapeutic proteins (monoclonal antibodies, cytokines, growth factors/hormones, enzymes, and fusion proteins) have made remarkable improvements for the treatment of numerous diseases, some of these products are associated with undesirable immunogenicity that may lead to reduced or loss of efficacy, altered pharmacokinetics (PK), neutralization of the natural counterpart, or even adverse clinical effects such as hypersensitivity reactions1,2.
In the context of therapeutic proteins, immunogenicity refers to the unwanted immune response of the host against the therapeutic protein; the immunogenic response usually generates anti-drug antibodies (ADAs) mediated by the activation of T-cells and subsequent stimulation of B-cells (Figure 1)3.
Immunogenicity occurs with uptake and processing of the therapeutic by dendritic cells or product-specific B cells which present a subset of its peptides on MHC-class II molecules on the cell surface. Recognition of MHC-class II by CD4+ T cells bearing T cell receptors (TCRs) stimulates T cell activation and cytokine production, with subsequent B cell activation. Post activation, differentiation into plasma B cells leads to the production and release of ADAs4. This process can take days to weeks after treatment and the immunogenicity effects vary depending on a combination of factors including patients, products, and treatment regimens3,5.
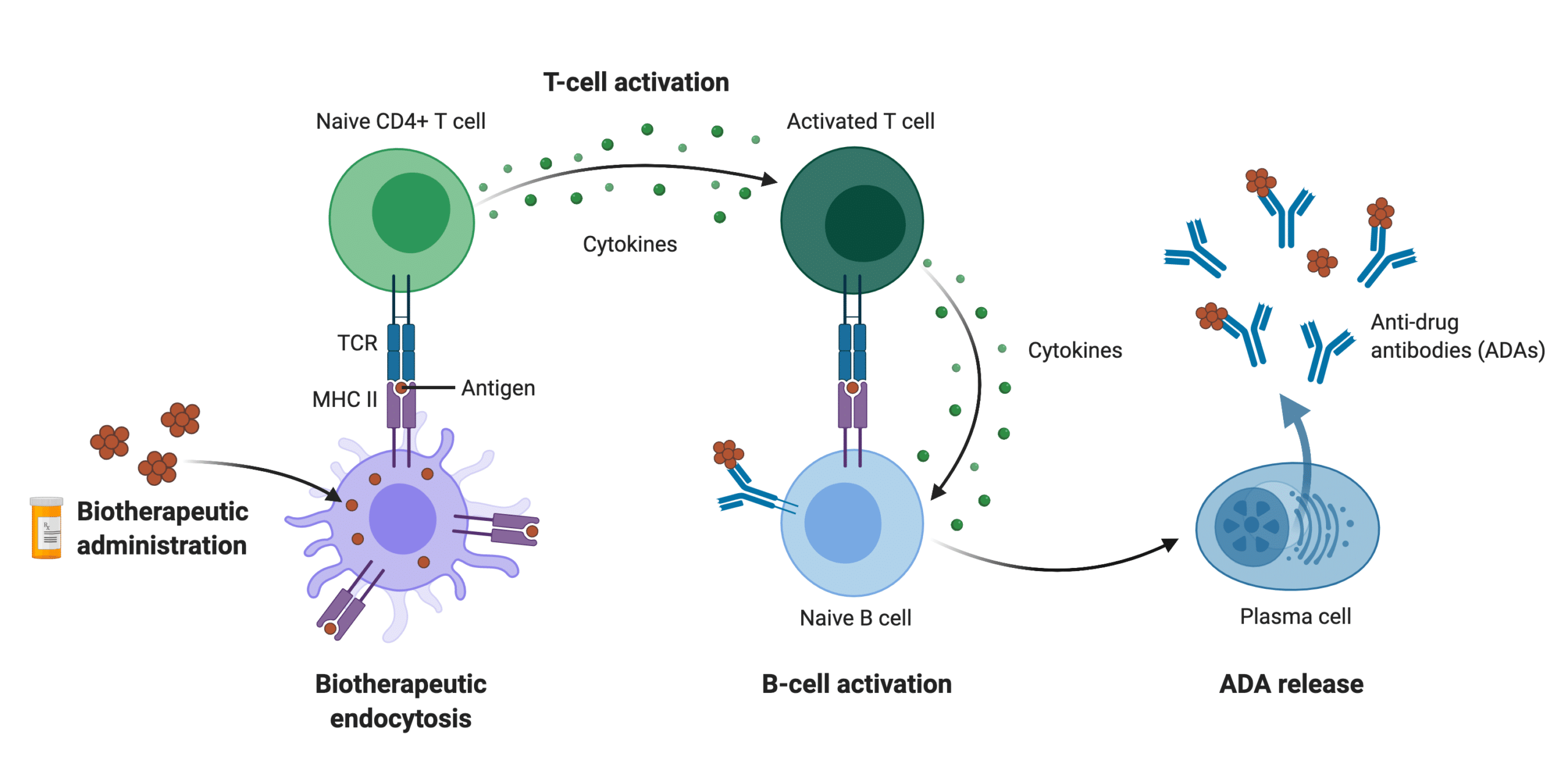
Figure 1. Schematic representation of the main immunogenic mechanism involved in biotherapeutic administration including the activation of the adaptive immune system via T-cell dependent priming of B cells to generate anti-drug antibodies.
Types of anti-drug antibodies
In practice, the clinical immunogenicity of therapeutics is measured and assessed by the detection of ADAs. ADAs are usually polyclonal, and may consist of different isotypes including IgM, IgG, IgE, and/or IgA6. Based on the immunogenicity mechanism caused by ADAs, ADAs can be classified into two types: binding antibodies or neutralizing antibodies (Figure 2).
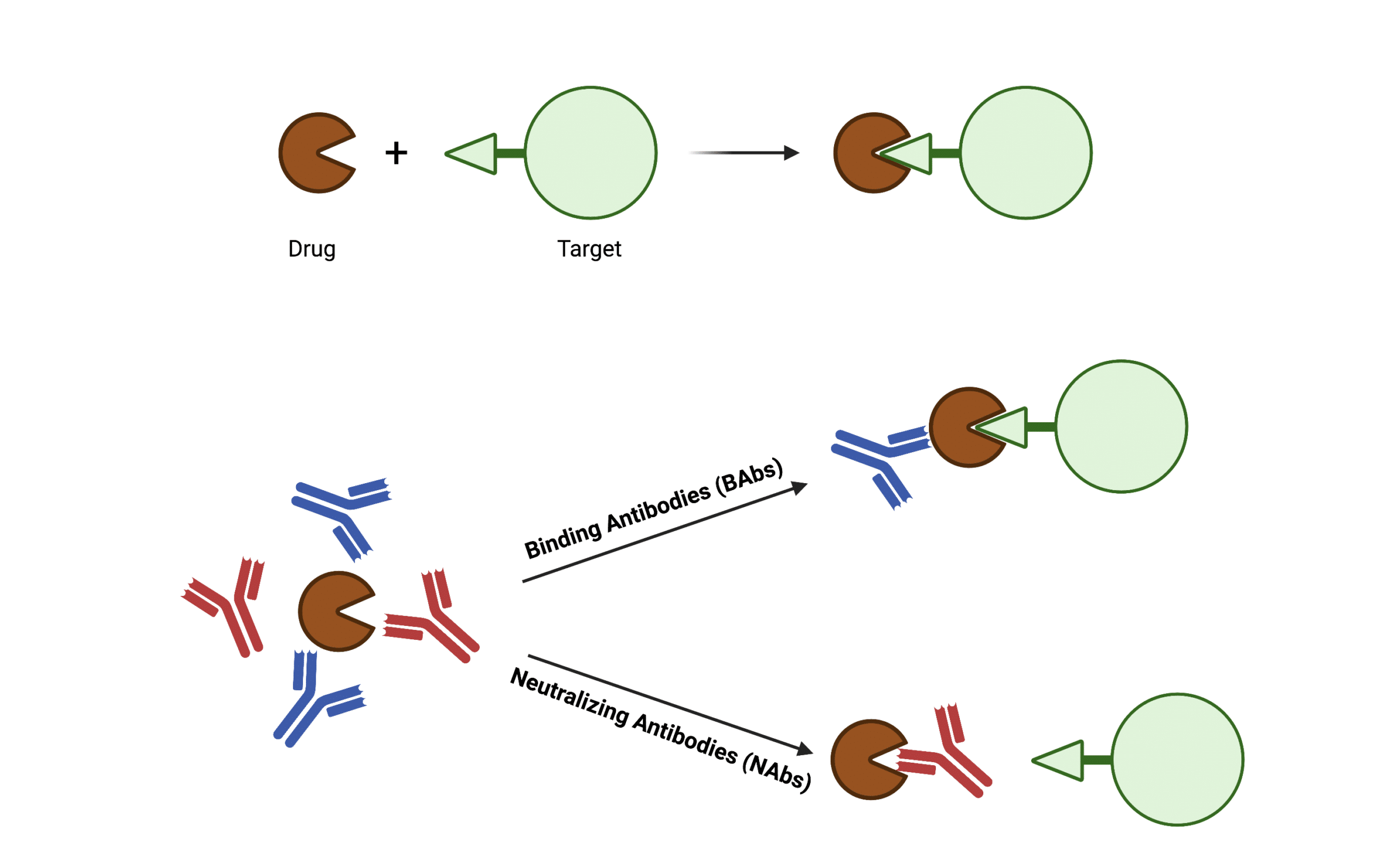
Figure 2. Drawing illustrating the two classifications of anti-drug antibodies: binding antibodies (BAbs) and neutralizing antibodies (NAbs) based on their impact on pharmacological drug function.
Binding antibodies (BAbs)
Binding antibodies (BAbs) bind to sites or epitopes of the therapeutic protein that do not participate in the interaction between the therapeutic and its target. BAbs are usually produced earlier during treatment in high amounts and persistency7,8. While BAbs do not necessarily impact the pharmaceutical activity of the therapeutic, they may either enhance the clearance of the therapeutic-ADA complex or affect the biological half-life of the administered agent9,10.
Neutralizing antibodies (NAbs)
Neutralizing antibodies (NAbs) are a subpopulation of the total ADAs that bind to the epitopes of therapeutic proteins responsible for target selectivity. By blocking the drug-target interaction, NAbs can inhibit the pharmacological function of the therapeutic, compromising clinical efficacy and provoking sometimes life-threatening allergic reactions11,12. Compared to BAbs, NAbs are produced later during treatment and tend to disappear or are markedly decreased in long-term treatment13.
Although NAbs are often of more concern in assessing therapeutic immunogenicity, both types of ADAs can have clinical effects such as poor efficiency and impaired safety14,15. Therefore, both NAbs and BAbs are clinically significant and immunogenicity risk assessment should be equally performed.
Examples of real-life ADA consequences
ADAs against coagulation factor VIII impair blood clotting
Coagulation factor VIII (FVIII) is an essential blood clotting protein and has been engineered as human recombinant protein used as a medication to treat hemophilia A. Unfortunately, inhibitory ADAs, or NAbs, against FVIII have been reported to develop in approximately 30% of treated patients16. Also, severe hemophilia A patients tend to acquire immune tolerance to FVIII during FVIII therapy, which negatively impacts clinical efficacy17. In some cases, production of inhibitory ADAs may cause severe clinical risks. For example, cross-reactive of ADAs with endogenous proteins results in thrombocytopenia17. Moreover, non-inhibitory binding ADAs can also contribute to uncontrolled bleeding due to increased FVIII clearance18.
NAbs in interferon α therapy can cause detrimental clinical effects
As a major family of anti-viral cytokines, interferon α (IFNα) in different recombinant forms has been made commercially available to treat chronic viral hepatitis and various cancers including hairy cell leukemia and malignant melanoma. Studies have reported that 19% – 61% of patients develop NAbs to IFNα-2a resulting in poorer IFNα treatment efficacy outcomes19. In a study of patients with chronic myelogenous leukemia treated with recombinant IFNα-2b, 30% generated NAbs and eventually suffered from a relapse or refractory response to IFN treatment20. Other adverse clinical effects in IFN treatment caused by immunogenicity include neutropenia, myelosuppression, anemia and thrombocytopenia2.
Current state of ADA assays
Overall approach: stepwise immunogenicity assessment
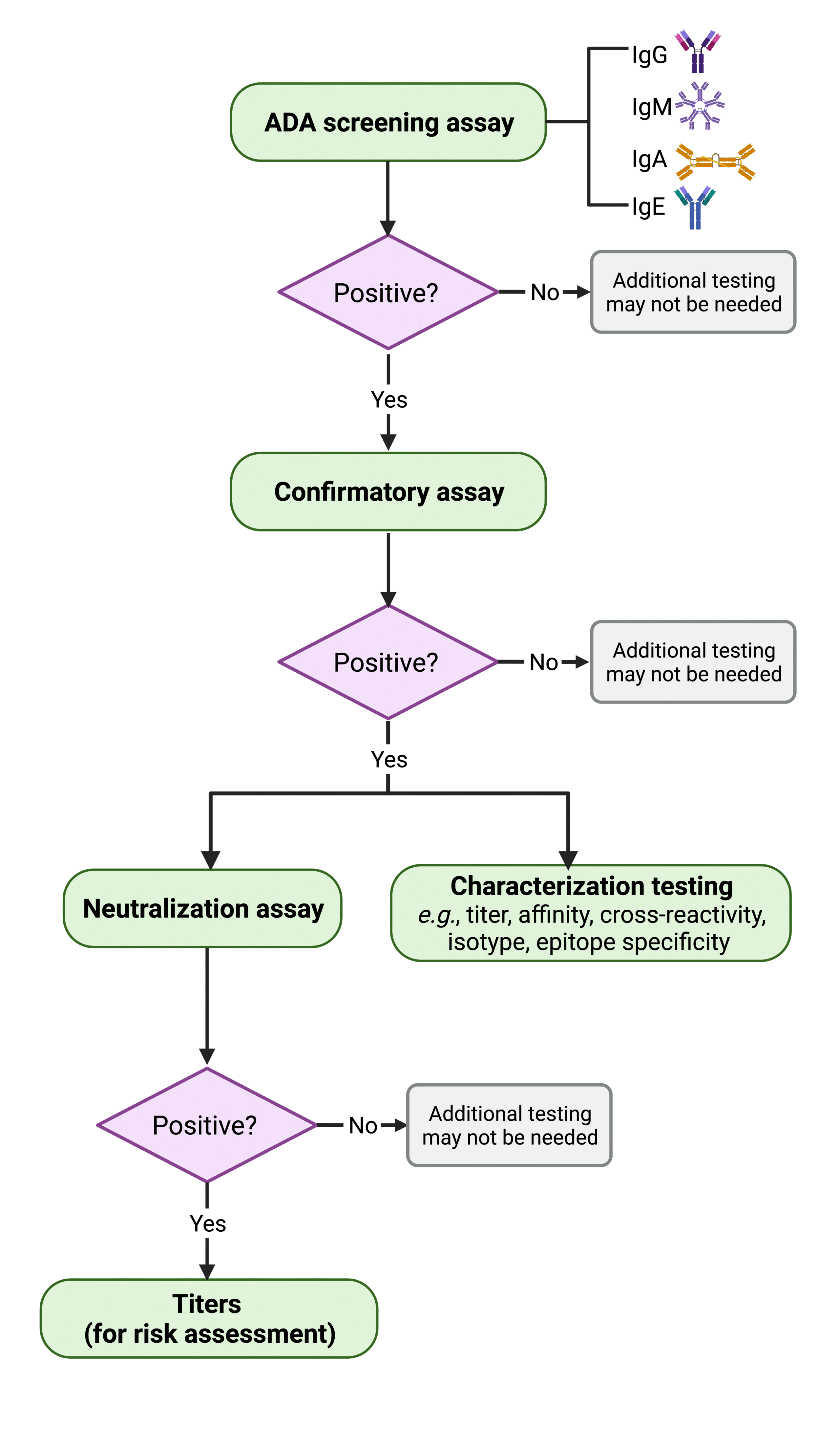
Figure 3. Flowchart depicting the steps involved in assessing biologicals and biosimilars’ immunogenicity. If immunogenicity is established, the ADAs are titrated to determine the likelihood of an adverse reaction, or the dose at which the drug loses efficacy modified from the FDA guidance of immunogenicity testing 2019 document21).
Development of appropriate assays to detect and measure ADAs is important for assessing immunogenicity of therapeutic protein products during both preclinical and clinical studies. A multi-tiered ADA testing approach has been recommended by the U.S. Food and Drug Administration (FDA) (Figure 3) and widely used by therapeutic developers21.
A complete ADA assessment process entails the following steps: First, a screening assay is employed to detect the presence of ADAs in all relevant isotypes in treated patients. Samples that tested as positive (found to contain ADAs) are also put through confirmatory assays to eliminate false positives and demonstrate that the ADAs present in the samples are specific to the therapeutic protein. Positives are further scrutinized with neutralization, titration, and other assays that characterize the biophysical properties (e.g., titer, isotype, cross-reactivity, epitope specificity, etc.) of each samples’ ADAs. Immunogenicity is assessed by considering all of the collected pharmacokinetics, pharmacodynamics, efficacy, and safety data.
ADA assay techniques
Several biochemical and bioanalytical techniques have been developed for applications in different stages of ADA testing. ADA screening and confirmatory assays include the enzyme-linked immunosorbent assay (ELISA), electrochemiluminescence (ECL) immune assay, radioimmunoassay (RIA), radioimmunoprecipitation assay (RIPA), and surface plasmon resonance (SPR). These tests have the advantages of robustness, sensitivity, and compatibility with readily available reagents and instruments. However, it is worth mentioning that the latter are only as robust as their standards.
Neutralization assays measure NAb activity through non-cell-based or cell-based experiments (Figure 4). The former (e.g., competitive ligand binding assays, and enzymatic activity assays) directly measure the binding between a surface-immobilized drug and target or vice versa. In contrast, cell-based bioassays have various outcomes such as phosphorylation of intracellular substrates, cell proliferation, cell death, or production of a secondary protein22. Therefore, cell-based assays tend to provide a more representative readout of the therapeutic’s in vivo mechanisms of action and are preferentially recommended by regulatory agencies such as the FDA21.
According to the FDA, an ideal ADA assay is expected to reliably detect ADAs with sensitivity, specificity, selectivity, precision, and reproducibility21. Thus, it is critical to implement assays with reliable, reproducible, and relevant standards, in addition to setting up the appropriate assay formats, and utilizing the right instruments for the job in order to meet regulatory requirements.
Positive control antibodies can make or break an ADA assay
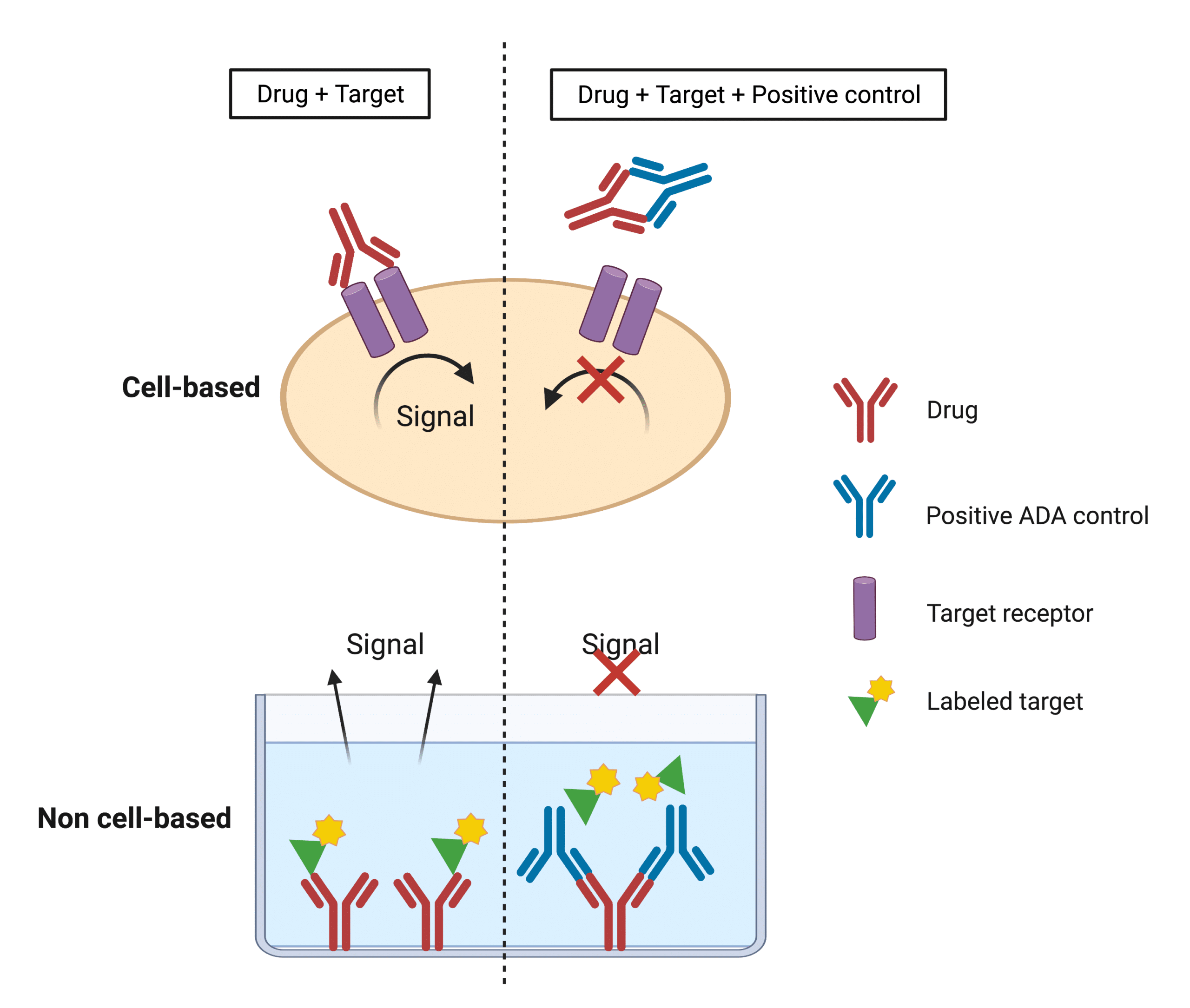
Figure 4. Illustration showing the basic principles of direct non-cell-based and cell-based assay formats to detect the presence of NAbs.
Positive control reagents that mimic the ADA response to the drug are indispensable to immunogenicity testing. These reagents typically consist of animal (e.g., rabbits)-derived pooled polyclonal antibodies (pAb) or human monoclonal antibody (mAb) reference panels against the target protein drug.
Such controls are important to determine tolerance and alerting to potential toxicity. But perhaps most importantly, they dictate the very validity and sensitivity of the assay. For instance, the FDA defines sensitivity of an ADA assay by the lowest concentration at which a positive control antibody consistently produces a positive result21. As such, positive control antibodies with higher affinity generally correlate with better sensitivity in contrast to lower affinity positive controls in the same assay.
To capture the complete immunogenic potential of a drug, an assay must assess the entire ADA response to said drug. But the latter can be difficult to accomplish when endogenously similar proteins exist, or when some of the drug remains in circulation. Such forms of interference lead to drug tolerance, which is defined in the lab as the maximum quantity of free drug that still results in a positive ADA readout. Beyond this limit, the assay may yield a negative readout that would falsely indicate the drug is not being bound by anti-drug antibodies (Figure 5).
Interference testing often requires different known amounts of positive control antibodies spiked into ADA-negative control samples in the absence or presence of different quantities of the protein therapeutic. Interference testing can thus be used to determine whether the biological drug will interfere with ADA detection21.
Unlike some other reagents and even assays that can be obtained from commercial sources (e.g., ELISA kits), control antibodies need to be tailored to each assay, and therefore require custom development. Due to the difficulty of acquiring enough human ADA for continued use, ‘surrogate’ positive controls are most frequently used. Such surrogates are pooled pAbs prepared from antiserum of immunized animals like rabbits. Alternatively, affinity-purified mouse or rat anti-human mAbs have been used6.
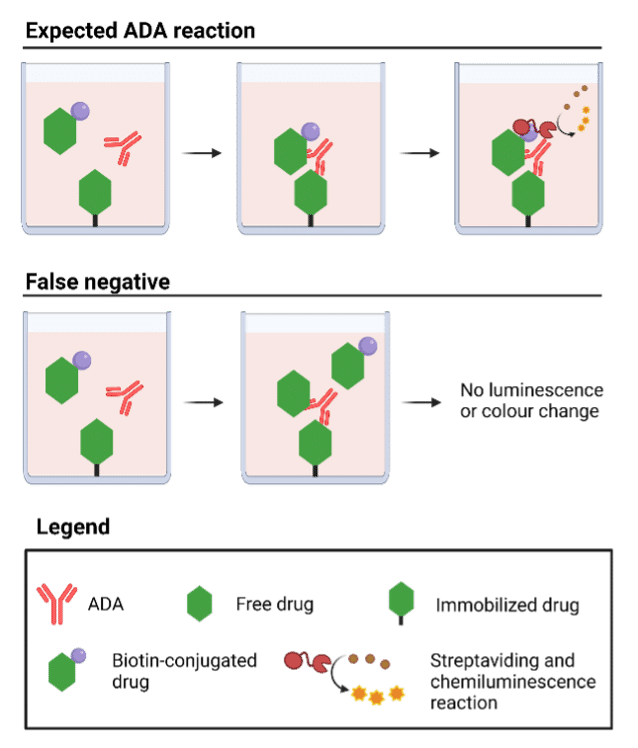
Figure 5. Illustration depicting how interference between free drug and ADA impedes proper interpretation of ADA assays – in this case, an ELISA.
Limitations of animal-derived positive controls
Animal-derived standards are prone to many challenges including issues in characterizing which epitope(s) is targeted by the ADA response as animal-derived and human ADAs typically have different binding patterns. Furthermore, due to the finite and variable nature of animal pAb production, new batches still require validation and characterization prior to clinical testing. And yet, use of individual mAbs may omit the inherent complexity of the ADA response, thus failing to provide clinically meaningful results.
Toward development of antibody reference panels
Another long-standing challenge of ADA assays is the lack of available relevant immunological reagents to the species being clinically assessed. The latter contributes to the paucity in ADA assay reproducibility that the scientific field has yet to overcome. In 2015, the first monoclonal antibody reference panel against recombinant human erythropoietin (EPO), a drug for treatment of anemia related to progressive kidney failure, was developed by the World Health Organization (WHO)23. A repertoire of nine human mAbs against human EPO were identified with defined characteristics including affinities (low to high), isotypes, epitopes, and neutralizing activities (available from NIBSC).
The development of this reference panel started with immunization of transgenic mice with human EPO. Upon serum titer screening, mice that tested positive were used for hybridoma generation, followed by further ELISA screening. Then hybridoma sequencing was performed to derive the sequence of each selected mAb for subsequent characterization24 (Figure 6). The performance of the antibody panel has been evaluated using different assays including ELISA, SPR, ECL, RIP, and biolayer-interferometry (BLI)23,25. Some platforms were able to detect the diverse repertoire of ADAs in the panel, clearly indicating the suitability and need for reference standards for improved performance in ADA assays.
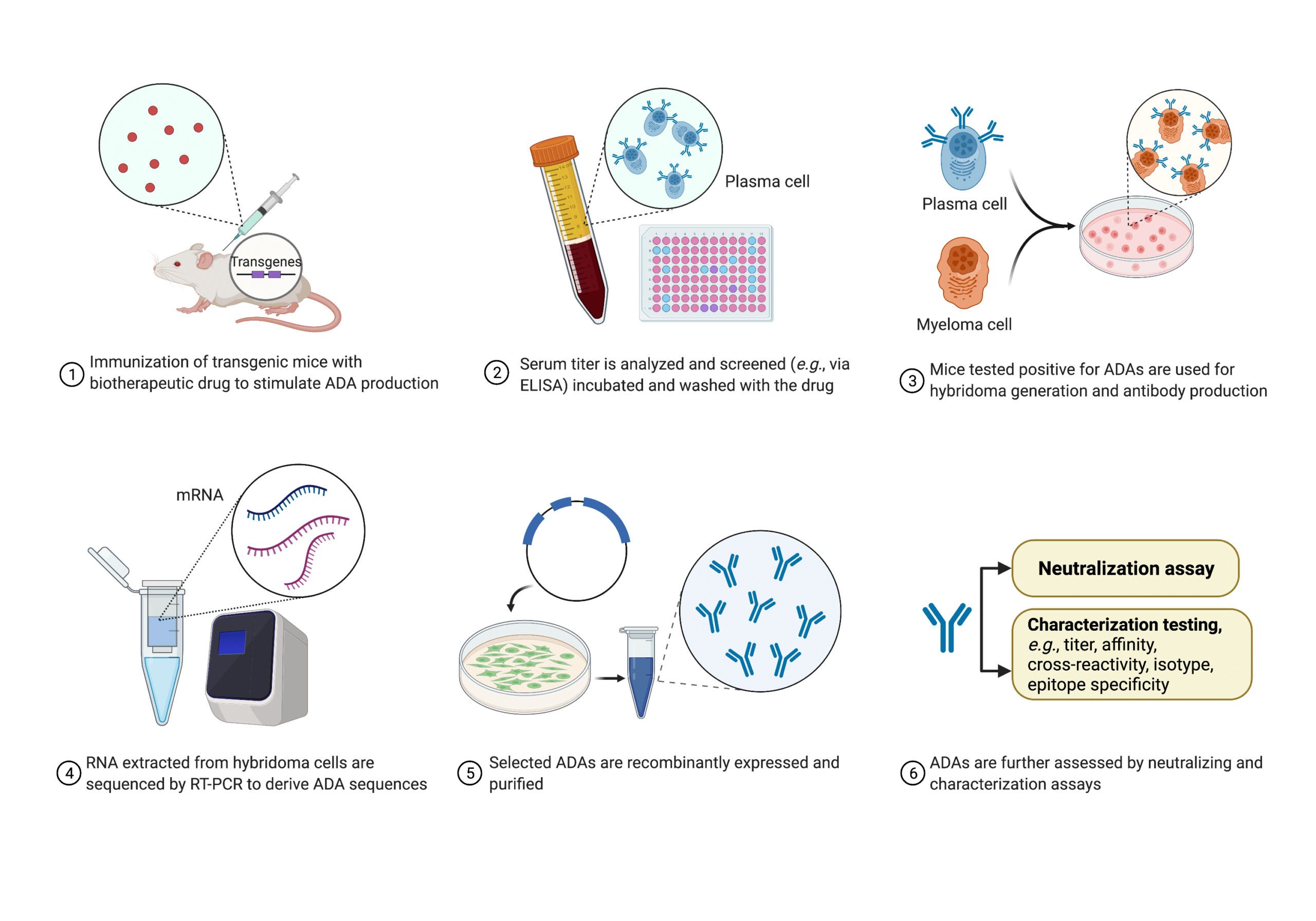
Figure 6. Process and techniques used by the WHO for developing antibody reference panels against EPO24.
Generating antibody reference panels presents an important step forward for the standardization of immunogenicity testing to facilitate the monitoring of the safety and efficacy of therapeutic protein products across different assay formats. Now, reference reagents, with established ranges, are recommended by the European Medicines Agency (EMA)26 and are under extensive development by clinical groups and regulatory agencies27.
Challenges of antibody reference panels
Despite the compelling benefits, human mAb reference panels can be tedious, costly, and time-consuming to generate using the conventional techniques such as aforementioned transgenic mice and hybridoma sequencing technologies. The repertoire of ADAs, which is polyclonal, needs to be detected and profiled to mimic the immune response to the drug as closely as possible. However, none of these techniques provides a direct and efficient tool to identify an adequate number of reference ADAs in an appropriate timeframe. For timely approval of a protein therapeutic, ADA assays should be developed and validated within 18 months of filing with the FDA. This can be a significant hurdle for many scientists in the industry.
Capturing the full ADA repertoire with next generation protein sequencing
Next generation protein sequencing is a term to coin the advances in protein sequencing in recent years. Protein sequencing refers to the elucidation of the amino acid order of a protein via liquid chromatography tandem mass spectrometric (LC-MS/MS) and bioinformatics-coupled analysis. Though widely published as peptide sequencing, very few laboratories around the world can accurately sequence proteins with this method because of the lack of sufficient mass spectrometric power, or poor bioinformatics development, either of which can seriously hamper sensitivity, protein coverage, and throughput. This is because congruous coupling of LC-MS/MS and bioinformatics is essential to ensuring the latter.
Overview of REpAb® polyclonal antibody sequencing
Pioneered by Rapid Novor, polyclonal antibody sequencing is an emerging technology that directly derives antibody sequences from blood or a pAb mixture. REpAb® polyclonal sequencing platform was built upon Rapid Novor’s well established monoclonal sequencing solution (REmAb®) that seamlessly integrates the latest advancements in mass spectrometry-based proteomics and big data bioinformatics. REpAb® is a powerful tool that can directly tap into the ADA repertoire by deriving the sequences of the polyclonal antibodies using only proteomics. However, though it speeds up developing ADA assay immunoreagents, it need not replace well-working, already-optimized pipelines. Certainly, REpAb® can complement transcriptomics data from hybridoma sequencing, or next generation sequencing (NGS). Regardless of the path chosen, REpAb’s technology has the capacity to mine more diverse and better characterized ADA immunoreagents for more reliable testing of a drug’s immunogenicity.
REpAb® workflow
REpAb® begins with the plasma or serum of an immunized animal or a patient after treatment, and two approaches are performed in parallel thereafter (Figure 7):
(1) REpAb® proteomics: Antigen-specific pAb mixture is affinity purified from the plasma for proteomics analysis, including protein digestion, LC-MS/MS, and machine learning-based sequence assembly. This generates antibody sequence data of both short peptides and intact proteins present in the mixture.
(2) REpAb® transcriptomics (optional): B cells are extracted from the plasma or serum and mRNAs of B-cell repertoire genes (BCRs) are sequenced by NGS to output transcriptomics data of the BCR. The latter often does not capture the full repertoire as many B cells are naïve, or few are circulating that produce the relevant antibodies. The spleen or bone marrow are very suited for this, but such protocols require culling of the production animal, or invasive medical procedures that are not ethically warranted for clinical drug testing. Even more so, relevant clones might still be missing during sorting or culturing. As such, tapping into the end product – the circulating antibodies – is critical to manufacturing more accurate and sensitive ADA positive controls.
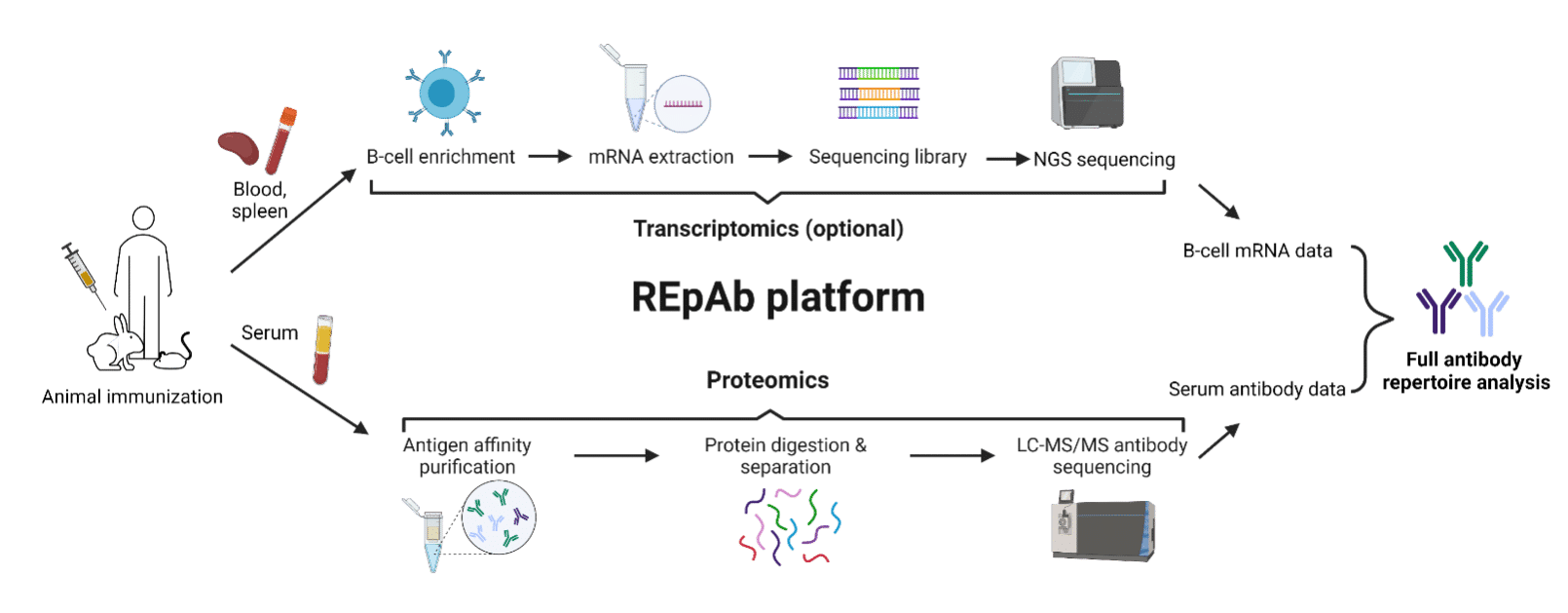
Figure 7. Flowchart providing an overview of REpAb® antibody discovery platform’s workflow.
Once both the proteomics and transcriptomics data are obtained, the two can be combined and cross-referenced to further assemble and derive the complete amino acid sequences of antibodies found in the pAb mixture. The sequences can then be used for in vitro recombinant expression to yield antibodies with higher reproducibility for an indefinite amount of time.
How REpAb® can facilitate generation of ADA antibody reference panels
REpAb®’s capability of decoding antibodies from blood makes it well suited to the development of antibody reference panels for ADA assays. Using REpAb®, protein sequences can be mined directly from the naturally occurring, patient polyclonal ADA response, bypassing time-consuming protocols such as transgenic animal model, and hybridoma generation. But when these platforms are already implemented, protein sequencing can provide additional relevant data such as post-translational modifications, and additional sequences that could have been missed during the nucleotide sequencing process (Figure 8).
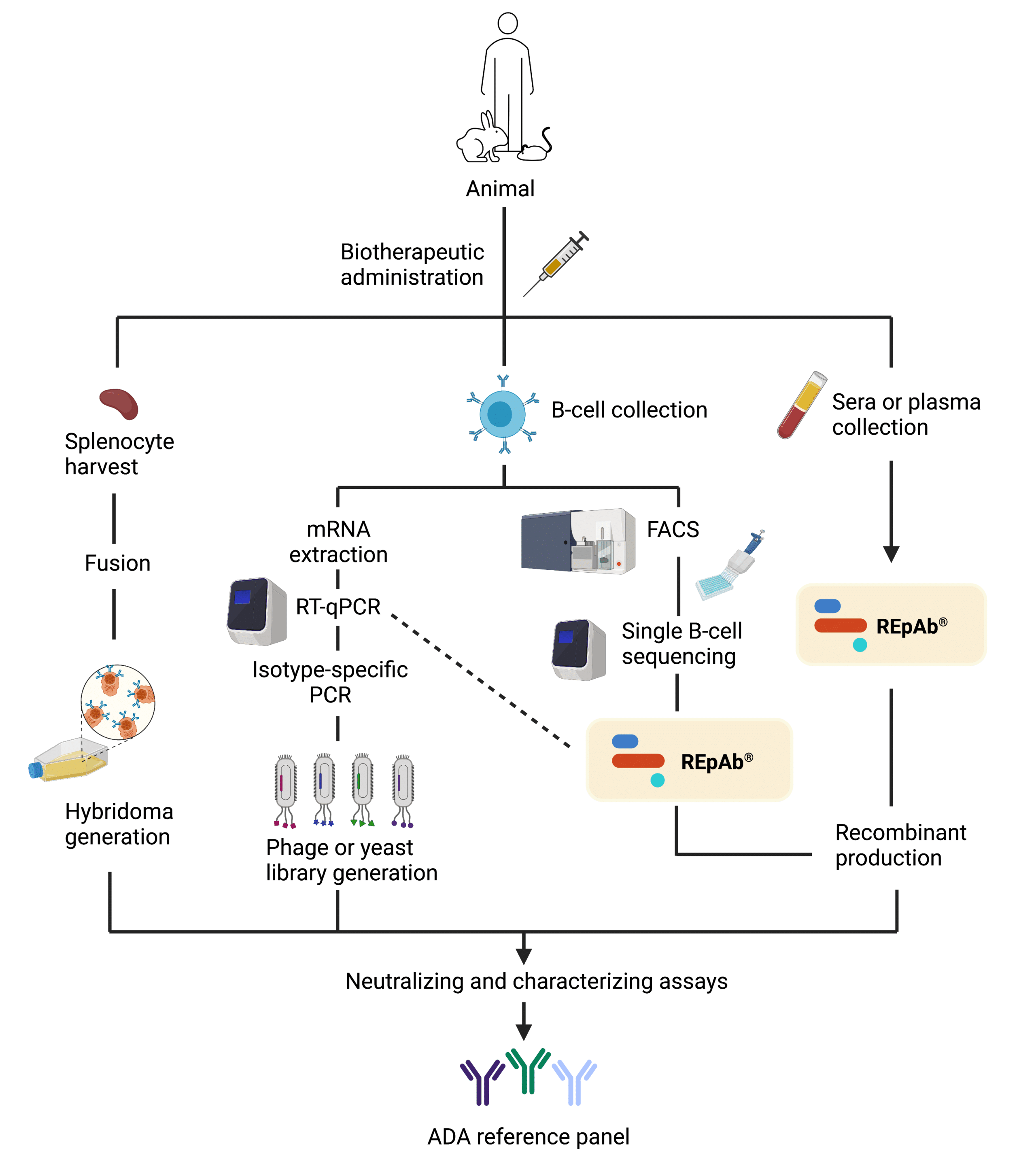
Figure 8. Schematic illustrating how REpAb® can complement other antibody discovery platform efforts in the generation of reference controls to facilitate and enhance ADA assay standardization and sensitivity, respectively.
Hence, direct access to the circulating antibody repertoire allows for a more comprehensive and accurate analysis of the humoral immune response. Therefore, such human-derived ADAs would be more strictly defined and bear high fidelity. Furthermore, because REpAb® is a de novo protein sequencing approach, it can sequence any isotype indiscriminate of the host species because it does not rely on databases of previously sequenced genomes. To sum up, with REpAb® technology, scientists can confidently rely on new and improved ADA positive controls to ease the characterization of ADA responses.
Conclusions
ADA assays need to meet stringent compliance requirements to ensure sensitivity and reliability. Identifying naturally occurring antibodies that reflect the patient immune response is the key to developing more sensitive ADA assays. REpAb® polyclonal sequencing technology is an attractive solution to profile the circulating ADA to finally completely capture immunogenic responses to biological drugs.
REpAb® advantages for ADA assays
- Accessible to all ADA isotypes (IgM, IgG, IgE, and IgA)
- Informative about PTMs such as glycosylation sites
- Most faithful representation of the patient ADA response
- Reduce time required to generate new ADA controls and “immortalize” standards for future testing
- Minimize use of laboratory animals, and cost of ADA development
Additional information
- Webinar: The New Era in Anti-Drug Antibody Assays
- pAb Sequencing for Next Generation Antibody Discovery
- Polyclonal Sequencing Using Only Proteomics
- Polyclonal sequencing in Antibody Discovery Pipeline
- Application Note: Biologics Drug Discovery
- Application Note: In vitro Diagnostics
- Application Note: Hybridoma Loss Prevention
References
- Büttel, I. C. et al. Taking immunogenicity assessment of therapeutic proteins to the next level. Biologicals 39, 100–109 (2011).
- Tovey, M. G. & Lallemand, C. Immunogenicity and other problems associated with the use of biopharmaceuticals. Ther Adv Drug Saf 2, 113–128 (2011).
- Gómez-Mantilla, J. D., Trocóniz, I. F., Parra-Guillén, Z. & Garrido, M. J. Review on modeling anti-antibody responses to monoclonal antibodies. J Pharmacokinet Pharmacodyn 41, 523–536 (2014).
- Rosenberg, A. S. & Sauna, Z. E. Immunogenicity assessment during the development of protein therapeutics. Journal of Pharmacy and Pharmacology 70, 584–594 (2018).
- Harding, F. A., Stickler, M. M., Razo, J. & DuBridge, R. The immunogenicity of humanized and fully human antibodies. mAbs 2, 256–265 (2010).
- Gunn, G. R. et al. From the bench to clinical practice: understanding the challenges and uncertainties in immunogenicity testing for biopharmaceuticals. Clin Exp Immunol 184, 137–146 (2016).
- Bellomi, F. et al. Fate of neutralizing and binding antibodies to IFN beta in MS patients treated with IFN beta for 6 years. J Neurol Sci 215, 3–8 (2003).
- Schellekens, H. Bioequivalence and the immunogenicity of biopharmaceuticals. Nat Rev Drug Discov 1, 457–462 (2002).
- Creeke, P. I. & Farrell, R. A. Clinical testing for neutralizing antibodies to interferon-β in multiple sclerosis. Ther Adv Neurol Disord 6, 3–17 (2013).
- Ponce, R. et al. Immunogenicity of biologically-derived therapeutics: Assessment and interpretation of nonclinical safety studies. Regulatory Toxicology and Pharmacology 54, 164–182 (2009).
- Pedotti, R. et al. An unexpected version of horror autotoxicus: anaphylactic shock to a self-peptide. Nat Immunol 2, 216–222 (2001).
- Rosenberg, A. S. Immunogenicity of biological therapeutics: a hierarchy of concerns. Dev Biol (Basel) 112, 15–21 (2003).
- Hegen, H. et al. Persistency of neutralizing antibodies depends on titer and interferon-beta preparation. Mult Scler 18, 610–615 (2012).
- Rojas, J. R. et al. Formation, distribution, and elimination of infliximab and anti-infliximab immune complexes in cynomolgus monkeys. J Pharmacol Exp Ther 313, 578–585 (2005).
- van der Laken, C. J. et al. Imaging and serum analysis of immune complex formation of radiolabelled infliximab and anti-infliximab in responders and non-responders to therapy for rheumatoid arthritis. Ann Rheum Dis 66, 253–256 (2007).
- Lusher, J. M. Inhibitor antibodies to factor VIII and factor IX: management. Semin Thromb Hemost 26, 179–188 (2000).
- Pratt, K. P. Engineering less immunogenic and antigenic FVIII proteins. Cell Immunol 301, 12–17 (2016).
- Batsuli, G. et al. High-affinity, noninhibitory pathogenic C1 domain antibodies are present in patients with hemophilia A and inhibitors. Blood 128, 2055–2067 (2016).
- Schellekens, H. & Casadevall, N. Immunogenicity of recombinant human proteins: causes and consequences. J Neurol 251 Suppl 2, II4-9 (2004).
- Freund, M. et al. Recombinant human interferon (IFN) alpha-2b in chronic myelogenous leukaemia: dose dependency of response and frequency of neutralizing anti-interferon antibodies. British Journal of Haematology 72, 350–356 (1989).
- Research, C. for D. E. and. Immunogenicity Testing of Therapeutic Protein Products —Developing and Validating Assays for Anti-Drug Antibody Detection. U.S. Food and Drug Administration https://www.fda.gov/regulatory-information/search-fda-guidance-documents/immunogenicity-testing-therapeutic-protein-products-developing-and-validating-assays-anti-drug (2020).
- Wu, B. et al. Strategies to Determine Assay Format for the Assessment of Neutralizing Antibody Responses to Biotherapeutics. AAPS J 18, 1335–1350 (2016).
- Wadhwa, M. et al. Establishment of the first WHO Erythropoietin antibody reference panel: Report of an international collaborative study. Journal of Immunological Methods 435, 32–42 (2016).
- Mytych, D. T. et al. Development and characterization of a human antibody reference panel against erythropoietin suitable for the standardization of ESA immunogenicity testing. Journal of Immunological Methods 382, 129–141 (2012).
- Finco, D. et al. Comparison of competitive ligand-binding assay and bioassay formats for the measurement of neutralizing antibodies to protein therapeutics. J Pharm Biomed Anal 54, 351–358 (2011).
- Anonymous. Immunogenicity assessment of biotechnology-derived therapeutic proteins. European Medicines Agency https://www.ema.europa.eu/en/immunogenicity-assessment-biotechnology-derived-therapeutic-proteins (2018).
- Wadhwa, M. & Thorpe, R. Harmonization and standardization of immunogenicity assessment of biotherapeutic products. Bioanalysis 11, 1593–1604 (2019)

Talk to Our Scientists.
We Have Sequenced 10,000+ Antibodies and We Are Eager to Help You.
Through next generation protein sequencing, Rapid Novor enables reliable discovery and development of novel reagents, diagnostics, and therapeutics. Thanks to our Next Generation Protein Sequencing and antibody discovery services, researchers have furthered thousands of projects, patented antibody therapeutics, and developed the first recombinant polyclonal antibody diagnostics.
Talk to Our Scientists.
We Have Sequenced 9000+ Antibodies and We Are Eager to Help You.
Through next generation protein sequencing, Rapid Novor enables timely and reliable discovery and development of novel reagents, diagnostics, and therapeutics. Thanks to our Next Generation Protein Sequencing and antibody discovery services, researchers have furthered thousands of projects, patented antibody therapeutics, and ran the first recombinant polyclonal antibody diagnostics